
Genetic Testing for Cystic Fibrosis - CAM 044
Description
Cystic fibrosis (CF) is an autosomal recessive genetic disease in which dysfunctional epithelial chloride channels lead to excessively thick mucus affecting multiple organ systems. Common complications include mucous plugging of the airway, lung inflammation, chronic pulmonary infections, intestinal malabsorption, pancreatic insufficiency, and infertility (Pritchard, 2016).
Policy
Application of coverage criteria is dependent upon an individual’s benefit coverage at the time of the request.
- Carrier screening for cystic fibrosis, using a panel containing mutations proven as causative of CF (as defined by the CFTR2 project) and including the ACMG’s 23 core mutations (see Policy Guideline #1 below), is considered MEDICALLY NECESSARY in the following situations:
- For all pregnant women OR
- For all women seeking pre-conception counseling OR
- For gamete donors OR
- For individuals who have a family history of cystic fibrosis or who have a first degree relative who is a known carrier of cystic fibrosis. (If not included in the panel, testing must also include any known familial mutations) OR
- For the male reproductive partners of women who have been identified as cystic fibrosis carriers OR
- For the reproductive partners of individuals diagnosed with cystic fibrosis
- Testing of a fetus for mutations in the CFTR gene (including all known parental mutations) is considered MEDICALLY NECESSARY when:
- Both biological parents are cystic fibrosis carriers
- One or both biological parents are affected with cystic fibrosis
- One biological parent is a cystic fibrosis carrier and the other parent is not available for testing
- Echogenic bowel is detected by fetal ultrasound
- Testing for mutations in the CFTR gene using a panel containing mutations proven as causative of CF (as defined by the CFTR2 project) and including the ACMG’s 23 core mutations (see Policy Guideline #1 below) is considered MEDICALLY NECESSARY in the following situations:
- In a newborn after an abnormal newborn screening result using immunoreactive trypsinogen, OR
- For infants with meconium ileus, OR
- For males with congenital absence of the vas deferens (CAVD), OR
- For individuals with idiopathic pancreatitis or bronchiectasis
- Testing for mutations in the CFTR gene is considered MEDICALLY NECESSARY as an adjunct to sweat testing in an individual presenting with symptoms of cystic fibrosis, in the following situations:
- When there are known familial mutations. Testing must include the familial mutations.
- When there are no known familial mutations, or if only one familial mutation is known. Testing can be done with a panel containing mutations proven as causative of CF (as defined by the CFTR2 project), including the ACMG’s 23 core mutations (see Policy Guideline #1 below), or testing can be done for the known mutation.
- Testing for mutations in the CFTR gene, using a panel containing mutations proven as causative of CF (as defined by the CFTR2 project) and including the ACMG’s 23 core mutations (see Policy Guideline #1 below), along with testing for the IVS8 5T/7T/9T variant, is considered MEDICALLY NECESSARY in males with congenital bilateral absence of the vas deferens (CBAVD).
- Testing for the IVS8 5T/7T/9T variant is considered MEDICALLY NECESSARY for cystic fibrosis carrier screening only as a reflex test when the R117H mutation is detected on carrier screening.
- When the clinical suspicion of cystic fibrosis remains in an individual for whom a panel containing mutations proven as causative of CF (as defined by the CFTR2 project), including the ACMG’s 23 core mutations (see Policy Guideline #1 below), came back with no mutations or only one mutation, comprehensive sequencing of the CFTR gene is considered MEDICALLY NECESSARY in the following conditions:
- In individuals that presented with symptoms of cystic fibrosis and who received panel testing as an adjunct to sweat testing.
- When a diagnosis of cystic fibrosis related CBAVD remains a consideration in males with CBAVD.
- Genetic counseling is considered MEDICALLY NECESSARY for:
- Individuals found to be cystic fibrosis carriers
- Individuals with a diagnosis of cystic fibrosis
- Individuals with a family history of cystic fibrosis
- Individuals who are the reproductive partner of a cystic fibrosis carrier
- Individuals who are the reproductive partner of a person diagnosed with cystic fibrosis or CBAVD
- Genetic testing for mutations in the CFTR gene is considered NOT MEDICALLY NECESSARY for all other indications.
Policy Guideline #1: A core panel of 23 mutations is recommended by the American College of Medical Genetics (ACMG) and is considered MEDICALLY NECESSARY for cystic fibrosis genetic testing. The standard mutation panel is as follows:
|
DF508 |
DI507 |
G542X |
G551D |
W1282X |
N1303K |
|
R553X |
621+1G®T |
R117H |
1717-1G®A |
A455E |
R560T |
|
R1162X |
G85E |
R334W |
R347P |
711+1G®T |
1898+1G®A |
|
2184delA |
3120+1G®A |
3849+10kbC®T |
2789+5G®A |
3659delC |
Rationale
Cystic fibrosis (CF) is a common life-limiting autosomal recessive genetic disorder caused by the mutation of a gene that encodes an epithelial chloride-conducting transmembrane channel called the cystic fibrosis transmembrane conductance regulator (CFTR). This gene regulates anion (negatively charged ion) transport and mucociliary clearance. Mucociliary clearance is the process by which particles and gases dissolved in the mucus are moved unidirectionally from the respiratory tract. The CFTR protein was first identified as a chloride channel but has been shown to facilitate or regulate the transport of other ions, such as sodium, thiocyanate, bicarbonate, as well as water absorption and excretion (Clancy & Jain, 2012; Cohen-Cymberknoh et al., 2011). The CFTR protein is present in the epithelia of various tissues, including that of the lungs, sweat glands, gastrointestinal tract, and pancreas (Barrett et al., 2012). CFTR dysfunction mainly affects epithelial cells; although, there is evidence of a role in immune cells.
Mutations in the CFTR gene which impede protein production, stability, or activity result in less available functional protein (Clancy & Jain, 2012). Functional failure of CFTR results in defective mucociliary clearance, chronic infection, and abnormal inflammatory response leading to progressive, irreversible lung damage (Cohen-Cymberknoh et al., 2011). Other manifestations of dysfunctional CFTR include pancreatic insufficiency and meconium ileus (Barrett et al., 2012). The early identification and treatment of patients by multidisciplinary teams have resulted in improvements in both quality of life and clinical outcomes in patients with cystic fibrosis, with life expectancy reaching 50 or even 60 years (Clancy & Jain, 2012).
Most mutations of the CFTR gene are missense alterations, but other mutations, deletions and insertions have been described (Bell et al., 2015). Missense mutations occur when a single nucleotide is changed, resulting in a different amino acid which may affect the overall protein functionality. To date, over 2000 mutations have been identified in the CFTR gene (CF Foundation, 2021). CFTR mutations can be divided into six classes according to their effects on protein function as depicted in the figure below (Bell et al., 2015).
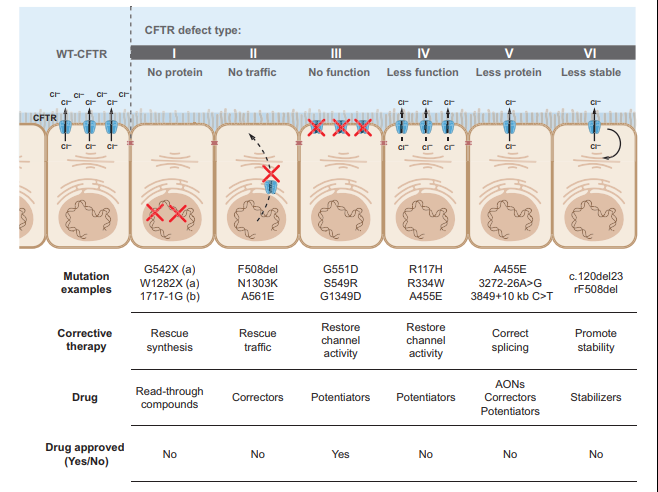
Class I, II, and III mutations are associated with no residual CFTR function and patients with these mutations have a severe phenotype, whereas individuals with class IV, V, and VI mutations have some residual function of CFTR protein and have a mild lung phenotype and pancreatic sufficiency (Bell et al., 2015; Wilschanski et al., 1995).
Due to the relatively high frequency in the U.S. population and studies demonstrating that early detection and care can improve outcomes, CF testing is included as part of the newborn screen in all states (Grosse et al., 2004). Newborn screening (NBS) for CF can include testing for immunoreactive trypsinogen (IRT), which is a pancreatic enzyme found at elevated levels in the blood of some individuals with CF, genetic testing for common CF mutations, or a combination of IRT and genetic testing. Positive newborn screens are typically followed by genetic testing (if not already done), and a sweat test, which measures the chloride concentration in sweat. The sweat test historically has been considered the “gold standard” for CF diagnosis. The presence of two disease-causing mutations also may be considered diagnostic of the disease, even in the absence of classic symptoms or in the case of a negative or inconclusive sweat test (Farrell et al., 2008).
In addition to NBS, CF carrier screening (PCS) has become commonplace in the U.S., particularly among pregnant women and couples planning a pregnancy. PCS has been found to markedly reduce CF birth rates with a shift towards milder mutations, but it was often avoided for cultural reasons necessitating the use of complementary PCS and NBS (Stafler et al., 2016).
A subset of 23 mutations account for the majority of cystic fibrosis cases in the U.S. and were accepted by most guidelines as the primary genes to be screened for diagnosis of CF and carrier status. The most common CF-causing mutation is F508del, which is present in over 70% of known CF cases. The Clinical and Functional Translation of CFTR (CFTR2) project continually evaluates genotype and phenotype correlations and has confirmed many additional mutations as being causative of CF (CF Foundation, 2021). Information from over 88,000 patients with specific cystic fibrosis variants from the United States, Canada, and Europe was collected by the CFTR2 team from national CF Patient Registries and placed in the CFTR2 database and then compiled into the CFTR2 website that contains information about the 322 most common CFTR variants (CF Foundation, 2021).
Analysis of data from the CF Foundation Patient Registry found that patients of Hispanic, black, or Asian ancestry were less likely to have two identified CFTR variants and more likely to carry no mutations on the commonly used 23 mutation carrier screening panel (Schrijver et al., 2016). Analysis of the Exome Aggregation Consortium dataset also found that none of the current genetic screening panels or existing CFTR mutation databases covered a majority of deleterious variants in any geographical population outside of Europe. Both clinical annotation and mutation coverage by commercially available targeted screening panels for CF are strongly biased toward detection of reproductive risk in persons of European descent (Lim et al., 2016). This research indicates the possible need for adjustment of this panel to facilitate equity in mutation detection between white and nonwhite or mixed-ethnicity CF patients, enabling an earlier diagnosis improving their quality of life.
Proprietary Testing
There are several cystic fibrosis genotyping assays approved by the FDA. eSensor Cystic Fibrosis Carrier Detection System detects 24 CFTR mutations (Clinical_Micro_Sensors, 2006), Tag-It™ Cystic Fibrosis Kit detects 39 mutations (Tm_Bioscience_Corporation, 2006), Cystic Fibrosis Genotyping Assay detects 30 mutations (Celera, 2007), InPlex™ CF Molecular Test detects 32 mutations (Hologic, 2008) , Verigene®CFTR and Verigene®CFTR PolyT Nucleic Acid Tests detect 26 mutations (Nanosphere, 2009), xTAG® Cystic Fibrosis 71 v2 Kit detects 71 mutations (Luminex, 2016) , and Illumina MiSeqDx™ Cystic Fibrosis 139-Variant Assay (Illumina, 2013).
Next generation sequencing (NGS) is also being integrated into CF diagnostic protocols. A diagnostic protocol in which a quick and relatively low-cost polymerase chain reaction (PCR)-based screening for the most common CF mutation, F508del, followed by full-gene next generation sequencing of CFTR was shown to improve genetic diagnosis and “holds promise to be a straightforward, convenient and rapid diagnostic protocol for CF patients” (Straniero et al., 2016). A study using NGS for NBS found that the NGS assay was 100% concordant with traditional methods. Retrospective analysis results indicate an IRT/NGS screening algorithm would enable high sensitivity, better specificity and positive predictive value (Baker et al., 2016). Further, whole genome sequencing of the CFTR gene is also growing in popularity. This type of sequencing has revealed a high prevalence of the intronic variant c.3874-4522A>G in those affected by CF (Morris-Rosendahl et al., 2020).
CFTR modulators are a new class of medications targeting the underlying defect in CF by improving production, intracellular processing, and/or function of the defective CFTR protein. Ivacaftor (IVA), which improves chloride channel function, IVA combined with lumacaftor (LUM), which partially corrects the CFTR misfolding, and IVA combined with tezacaftor, which improves the intracellular processing and trafficking of CFTR, have been approved by the U.S. Food and Drug Administration for use in patients with CF. The indications and efficacy of these drugs depend upon the CFTR mutation in the individual patient. For example, ivacaftor has been FDA-approved and Clinical Pharmacogenetics Implementation Consortium (CPIC)-recommended only for CF patients with the G551D mutation. It is not recommended for any other CF mutation (Clancy et al., 2014).
Analytical Validity
Lyon et al. (2014) assessed clinical laboratories’ proficiency at evaluating genetic alterations for CF. A total of 357 labs participated, performing approximately 120,000 tests monthly. Analytical sensitivity and specificity were 98.8% and 99.6%, respectively. Clinical interpretation matched intended response for zero, one, and two mutations. The authors concluded that laboratory testing for CF in the United States is of high quality (Lyon et al., 2014).
Sugunaraj et al. (2019) completed a cross-sectional study which included the analysis of CFTR variants in 50,778 exomes. Only 24 patients were identified to contain bi-allelic pathogenic CFTR variants; the authors identified 21 of these cases as true-positives and three as potential false positives. Therefore, “genomic screening exhibited a positive predictive value of 87.5%, negative predictive value of 99.9%, sensitivity of 95.5%, and a specificity of 99.9%” (Sugunaraj et al., 2019). Overall, the presence and/or absence of CFTR variants was strongly related to a CF diagnosis.
Hendrix et al. (2020) assessed the performance of dried blood spot DNA extraction methods in NGS for cystic fibrosis. 20 DNA samples were used in this study and sequenced using Illumina's MiSeqDx™ Cystic Fibrosis 139-Variant Assay and Swift Biosciences’ Accel-Amplicon™ CFTR panel. The MiSeqDx CFTR Clinical Sequencing Assay identified 37 CF-causing variants in the 20 samples tested, for a total of 141 benign variants which were all confirmed with Sanger sequencing. The Swift Biosciences’ Accel-Amplicon™ CFTR panel identified 39 CF-causing variants for a total of 123 benign variants which were all confirmed with Sanger sequencing. Illumina's MiSeqDx performed well with a very high average coverage per method (> 3000×) despite most DNA extraction methods being low-concentration and relatively crude. Because of the higher input of samples on a single flow cell, the average coverage per target was lower in the Accel-Amplicon™ CFTR panel in comparison to the clinical sequencing assay, but still often had >1000× coverage. The authors conclude that “both these tests are appropriate DNA extraction methods that can be used with dried blood spots that will result in consistent, high-quality sequencing results” (Hendrix et al., 2020).
Sicko et al. (2021) validated the use of a custom NGS assay, Archer CF assay, for CF newborn screening. According to the New York State, CF NBS had high false positive rates. However, after the implementation of this three-tier IRT-DNA-SEQ approach using commercially available tests, the positive predictive value of CF NBS improved from 3.7% to 25.2%. The Archer CF assay involves full gene sequencing, analyzing a specific panel of 338 clinically relevant CFTR variants (second tier), followed by unblinding of all sequence variants and bioinformatic assessment of deletions/duplications (third tier). 227 infant samples were taken, and 190 of 227 carries one or more of the 338 panel variants. The second-tier panel variants were all called correctly, detecting 44 different variants with a 100% sensitivity and 100% specificity. All 227 samples also underwent third tier analysis which detected 62 additional unique SNV/indel variants (Sicko et al., 2021).
Clinical Utility and Validity
Sosnay et al. (2013) performed a comprehensive analysis of CF genotypes and phenotypes. The authors collected genotype and phenotype data from 39696 CF patients and found 159 variants with over 0.01% allele frequency, 127 of which met both the clinical and functional criteria for CF. The phenotypic criterion used to define the pathogenic threshold was sweat chloride conductance of 10%. These 127 pathogenic genetic variants were estimated to represent 95.4% of CF alleles, leaving only 0.21% of CF patients without an identified pathological CFTR variant. The phenotypes of these variants vary (Sosnay et al., 2013).
Wainwright et al. (2015) evaluated the combination of IVA and LUM in CF patients homozygous for the F508D mutation (labeled Phe508del). A total of 1108 patients were examined. The mean baseline forced expiratory volume in one second “FEV1” was 61% of the predicted value. The absolute mean improvement in FEV1% was found to be 2.6-4%, which corresponded to a 4.3-6.7% relative treatment difference. The rate of pulmonary exacerbations was found to be 30-39% less in the treatment group compared to the placebo group. However, the rate of discontinuation due to an adverse event was 4.2% in the treatment group compared to the placebo group (Wainwright et al., 2015).
Sharma et al. (2018) performed a cost-effectiveness analysis of the FDA-approved IVA-LUM combination for the F508D mutation. The authors built a Markov-state model to assess the IVA-LUM for 12-year-old CF patients over several periods of time. “Markov states included mild CF (percentage of predicted forced expiratory volume in 1 second or FEV1 > 70%), moderate (FEV1 40–70%), severe (FEV1 < 40%) disease, post-transplant, and death. Pulmonary exacerbation and lung transplant were included as transition states.” The IVA-LUM combination resulted in higher quality adjusted life years (QALY, 7.29 vs 6.84 for usual care) but at a cost of $1,778,920.88 per QALY compared to $116,155.76 for usual care. Both monetary amounts were over a 10-year period. The IVA-LUM combination was cost-effective at a threshold of $150,000 / QALY, which was estimated to occur at an annual drug cost of $4153 (Sharma et al., 2018).
Kessels et al. (2020) completed a systematic review of prenatal genetic testing for CF. Eight different databases were researched for this review. The authors found that after genetic testing “A change in clinical management was observed: termination of pregnancy (TOP) occurred in most cases where two pathogenic variants were identified in a fetus of carrier parents (158/167; 94.6%)” (Kessels et al., 2020). The authors conclude by stating that genetic testing for CF had good diagnostic performance and lead to fewer births affected by CF.
Avram et al. (2021) studied the cost-effectiveness of genotyping versus sequencing for prenatal CF carrier screening using 3 strategies: genotyping (“genotyping included the standard 23-variant panel recommended by ACMG/ACOG”) both partners, genotyping one partner then sequencing the second, and sequencing both partners. The authors used a decision-analytic model to generate a theoretical cohort of four million pregnant people and their partners. Sequencing both partners identified 1,099 carrier couples that would have been missed by genotyping of both partners, resulting in 273 fewer missed prenatal diagnoses, 152 more terminations, 152 fewer affected newborns, and screening with genotyping followed by sequencing identified 477 more carrier couples as compared to genotyping both partners, resulting in 119 fewer missed prenatal diagnoses, 66 more terminations, and 66 fewer affected newborns. The incremental cost-effectiveness ratio of genotyping followed by sequencing compared to genotyping both partners was $180,004/ quality-adjusted life year (QALY) and the incremental cost-effectiveness ratio of sequencing both partners compared to genotyping followed by sequencing was $17.6 million/QALY. “Compared to genotyping both partners, the cost per CF case averted for genotyping/sequencing was $1.2 million and sequencing both partners was $49.4 million .... Sequencing both partners was cost-effective when the cost of sequencing fell below $339 per test. Genotyping/sequencing was cost-effective when the cost of sequencing was between $340 and 1837 per test. Lastly, genotyping both partners was cost-effective when sequencing cost above $1838 per test.". The authors concluded that “before being routinely recommended, sequencing for CF carrier screening requires greater examination at a population-based level (Avram et al., 2021).
Cystic Fibrosis Foundation
The Cystic Fibrosis Foundation (Farrell et al., 2017) convened a group of to develop clear and actionable consensus guidelines on the diagnosis of CF and to clarify diagnostic criteria and terminology for other disorders associated with CFTR mutations. The experts determined that “diagnoses associated with CFTR mutations in all individuals, from newborn to adult, be established by evaluation of CFTR function with a sweat chloride test. The latest mutation classifications annotated in the Clinical and Functional Translation of CFTR project (http://www.cftr2.org/index.php) should be used to aid in diagnosis.”
The committee approved 27 consensus statements (Farrell et al., 2017):
- Sweat chloride testing should be performed according to approved procedural guidelines published in established, international protocols such as the CLSI 2009 Guidelines.
- Newborns with a positive CF newborn screen, to increase the likelihood of collecting an adequate sweat specimen, should have the test performed bilaterally and when the infant weighs > 2 kg, and is at least 36 wk of corrected gestational age.
- Newborns greater than 36 wk gestation and >2 kg body weight with a positive CF newborn screen, or positive prenatal genetic test, should have sweat chloride testing performed as soon as possible after 10 d of age, ideally by the end of the neonatal period (4 wk of age).
- In infants with presumptive CF identified through NBS, CF treatment should not be delayed while efforts to establish a diagnosis of CF are initiated.
- Sweat chloride analysis should be performed within a few hours of sweat collection and the results and interpretations should be reported to clinicians and parents or patients, as soon as possible and certainly on the same day.
- In individuals presenting with a positive newborn screen, clinical features consistent with CF, or a positive family history, a diagnosis of CF can be made if the sweat chloride value is ≥ 60 mmol/L.
- Individuals who are screen-positive and meet sweat chloride criteria for CF diagnosis should undergo CFTR genetic testing if the CFTR genotype was not available through the screening process or is incomplete.
- In individuals with a positive newborn screen, a sweat chloride < 30 mmol/L indicates that CF is unlikely.
- Individuals with clinical features that may be consistent with CF who have a sweat chloride < 30 mmol/L indicates that CF is less likely. It may, however, be considered if evolving clinical criteria and/or CFTR genotyping support CF and not an alternative diagnosis.
- Individuals presenting with a positive newborn screen, symptoms of CF, or a positive family history, and sweat chloride values in the intermediate range (30 – 59 mmol/L) on two separate occasions may have CF. They should be considered for extended CFTR gene analysis and/or CFTR functional analysis.
- The latest classifications identified in the CFTR2 project (http://www.cftr2.org/index.php) should be used to aid with CF diagnosis:
- CF-causing mutation: individuals with 2 copies on separate alleles will likely have CF (clinical sweat confirmation needed)
- Mutation of varying clinical consequence (MVCC): a mutation that in combination with a CF-causing mutation or another MVCC mutation may result in CF
- Uncharacterized mutation/mutation of UNK: mutation that has not been evaluated by CFTR2 and may be disease causing or of variable clinical consequence or benign
- Non-CF-causing mutation: individuals with 1 or more are unlikely to have CF (as a result of that allele)
- In individuals presenting with a positive newborn screen, symptoms of CF, or a positive family history, the identification of 2 CF-causing mutations (defined by CFTR2) is consistent with a diagnosis of CF. Sweat chloride testing is necessary, though, to confirm the diagnosis.
- The absence of detection of 2 CF-causing CFTR mutations does not exclude a diagnosis of CF.
- If further CF functional testing is needed (NPD and ICM), it should be performed in a validated reference center with trained staff certified by the CF Foundation TDN or ECFS Clinical Trial Network.
- In individuals with a positive newborn screen but variable or uncharacterized CFTR mutations (< 2 CF-causing mutations), the diagnosis of CF can be made by demonstrating CFTR dysfunction (a sweat chloride ≥ 60 mmol/L or CF-typical NPD or ICM).
- The term CRMS is used in the US for healthcare delivery purposes and CFSPID is used in other countries, but these both describe an inconclusive diagnosis following NBS.
- The term CRMS/CFSPID is reserved for individuals who screen positive without clinical features consistent with a diagnosis of CF.
- The definition of CRMS/CFSPID is an infant with a positive NBS test for CF and either:
- A sweat chloride value <30 mmol/L and 2 CFTR mutations, at least 1 of which has unclear phenotypic consequences OR
- An intermediate sweat chloride value (30-59 mmol/L) and 1 or 0 CF-causing mutations
- Children designated as CRMS/CFSPID should undergo at least one repeat sweat chloride test at CF centers with suitable expertise, such as an accredited CF center.
- Children designated as CRMS/CFSPID should have clinical evaluation performed by CF providers to identify the minority that may develop clinical symptoms.
- Children designated as CRMS/CFSPID can be considered for extended CFTR gene analysis (sequencing and or deletion duplication testing), as well as CFTR functional analysis (NPD/ICM) testing to further define their likelihood of developing CF.
- The decision to reclassify children designated as CRMS/CFSPID as CF is an integrated decision that should take into account functional assessment of CFTR (sweat chloride, and possibly NPD/ICM), CFTR genetic analysis, and clinical assessment by the CF clinicians caring for the patient.
- Genetic counseling should be offered to families of individuals followed for CRMS/CFSPID, including a discussion of the risk in future pregnancies.
- Research Recommendation: Infants with a designation of CRMS/CFSPID (by definition) do not have clinical features consistent with a diagnosis of CF and further research is needed to determine the prognosis and best practices for frequency and duration of follow-up.
- For individuals presenting with CF symptoms, the same diagnostic criteria recommended for the screened population for sweat chloride testing, CFTR genetic analysis, and CFTR functional testing should be used to confirm a CF diagnosis.
- The diagnosis of CFTR-related disorder has been defined as a monosymptomatic clinical entity (CBAVD/pancreatitis/bronchiectasis) associated with CFTR dysfunction that does not fulfill the diagnostic criteria for CF.
- Clinicians should avoid the use of terms like classic/nonclassic CF, typical/atypical CF, delayed CF, because these terms have no harmonized definition and could be confusing for families or caregivers.
The Cystic Fibrosis Foundation (Ren et al., 2018) also convened a multidisciplinary committee of CF caregivers to develop evidence-based guidelines for CFTR modulator therapy. The committee defined patients with CF as individuals who met the above CFF criteria for diagnosis of CF, combined with evidence of abnormal CFTR function, as demonstrated by elevated sweat chloride, detection of two CF-causing CFTR mutations, or abnormal nasal potential differences.
For adults and children aged 6 years and older with CF due to gating mutations other than G551D or R117H, the guideline panel made a conditional recommendation for treatment with IVA. For those with the R117H mutation, the guideline panel made a conditional recommendation for treatment with IVA for 1) adults aged 18 years or older, and 2) children aged 6–17 years with a forced expiratory volume in 1 second (FEV1) less than 90% predicted. For those with the R117H mutation, the guideline panel made a conditional recommendation against treatment with IVA for 1) children aged 12 – 17 years with an FEV1 greater than 90% predicted, and 2) children less than 6 years of age. Among those with two copies of F508del, the guideline panel made a strong recommendation for treatment with IVA/LUM for adults and children aged 12 years and older with an FEV1 less than 90% predicted; and made a conditional recommendation for treatment with IVA/LUM for 1) adults and children aged 12 years or older with an FEV1 greater than 90% predicted, and 2) children aged 6 – 11 years.
American College of Obstetrics and Gynecology (ACOG)
The ACOG recommends offering CF carrier screening to all women who are considering pregnancy or are currently pregnant. Expanded mutation panels which enhance the sensitivity for carrier screening can be considered for carrier detection, especially in non-Caucasian ethnic groups, but testing should not be repeated if performed previously (e.g., during previous pregnancy). If the patient is found to be a carrier, then her partner should be tested. They also indicate that newborn screening is not a replacement for carrier screening in a population.
Prenatal diagnosis is indicated after genetic counseling if both parents are carriers, or if the mother is a carrier and the father is unknown or unavailable for testing.
ACOG recommends that complete gene sequencing of the CTFR gene is not appropriate for use in carrier screening, rather reserved for patients with cystic fibrosis, patients with negative carrier screening but a family history of cystic fibrosis, males with congenital bilateral absence of the vas deferens, or newborns with a positive newborn screening after the standard 23 gene screen has a negative result.
They also recommend referral to genetic counseling for couples in which both partners are CF carriers, prenatal diagnosis and advanced reproductive technologies to decrease the risk of affected offspring should be discussed (ACOG, 2017).
These statements were reaffirmed in 2020.
American College of Medical Genetics and Genomics (ACMG)
The ACMG also recommends offering CF carrier screening to all women of reproductive age, regardless of ethnic background. Ideally, the testing would occur prior to pregnancy, but if the woman is pregnant, then she should be tested as early as possible during the pregnancy. ACMG also indicates that if the woman is found to be a CF carrier, then her reproductive partner should also be tested (Pletcher & Bocian, 2006). If the woman presents for testing late in the pregnancy, simultaneous testing of both the woman and the biologic father of the fetus should be considered. When there is a paternal family history of CF, carrier testing of the father is warranted.
Additionally, ACMG defines a panel of 23 mutations that includes the most common mutations found in a pan-ethnic population in the U.S. for use in routine CF carrier screening (ACMG, 2011; Watson et al., 2004). This 23-mutation panel incorporates all CF-causing mutations with a frequency of greater than or equal to 0.1% in the general U.S. population. ACOG also endorses the ACMG panel, and both organizations recommend against use of expanded mutation panels for routine CF carrier screening.
ACMG also recommends that testing for the IVS8 polyT variant be performed only when carrier screening reveals a R117H mutation, as the polyT variant influences the clinical severity of the mutation but does not cause CF by itself (ACMG, 2011).
ACMG provides guidance on indications for CFTR variant testing. Diagnostic testing can be used for the molecular confirmation of a clinical CF diagnosis, for infants with meconium ileus, for males with CAVD, individuals with idiopathic pancreatitis or bronchiectasis, and as a follow-up to newborn screening. Regarding carrier testing, ACMG recommends that carrier testing be offered to “individuals with a positive family history of CF, in partners of individuals with a positive family history, in partners of CAVD males, to reproductive age women, and to gamete donors” (Deignan et al., 2020).
Prenatal testing can be performed using cells obtained for diagnostic cytogenetic testing (i.e., amniocentesis or chorionic villus sampling [CVS]). Testing can take place on cultured or uncultured amniocytes or villous trophoblasts. According to the guidelines, targeted sequencing for specific CFTR variants may be considered when:
- “A pathogenic or likely pathogenic variant is confirmed in both partners.
- A pathogenic or likely pathogenic variant is confirmed in one partner and a VUS or variant associated with variable expressivity is confirmed in the other partner
- As part of preimplantation genetic testing when both biological parents are confirmed carriers of a pathogenic or likely pathogenic variant” (Deignan et al., 2020).
Comprehensive CFTR sequencing may be considered when:
- “One member of a couple is known to be a carrier of a pathogenic or likely pathogenic variant and any of the following is also true:
- The partner is unavailable for screening
- The partner has not been screened and any of the following is also true:
- Screening that partner would be cost prohibitive
- The results from the partner would not be available in time to allow for reproductive decision making
- A diagnostic procedure (e.g., CVS, amniocentesis) is also being performed for other reasons (e.g., ultrasound abnormality).
- An ultrasound finding (i.e., fetal echogenic bowel) suggests an affected fetus and CFTR variant information is not available from either biological parent” (Deignan et al., 2020).
ACMG recommends the following regarding testing of intronic variants, deletions/duplications, reporting VUS and variable expressivity, and reporting of intron 9 polyT and TG regions:
- “ACMG recommends the reporting of the c.2657+5G>A and c.3718–2477C>T intronic variants. If these variants are not detectable with the laboratory’s methodology (e.g., exome sequencing), then a separate assay should be performed to test for these variants.
- For all prenatal, postnatal, and adult diagnostic testing and carrier screening indications for CFTR testing, the ACMG does not recommend the testing of any specific exon-level or gene-level deletion or duplication variants.
- For all prenatal, postnatal, and adult diagnostic testing indications for CFTR where comprehensive methods are used, the ACMG recommends the reporting of VUS. For all adult carrier screening indications for CFTR where comprehensive methods are used, VUS should generally not be reported. However, laboratories may want to consider reporting VUS in the partner of an individual who had a pathogenic or likely pathogenic variant detected during screening.
- For all prenatal, postnatal, and adult diagnostic testing indications for CFTR where comprehensive methods are used, the ACMG recommends the reporting of any variants associated with variable expressivity.
- For all prenatal, postnatal, and adult diagnostic testing indications for CFTR, the ACMG recommends the reporting of R117H status as well as the results from at least the associated polyT tract. For all adult carrier screening indications for CFTR, polyT status should be reported when the R117H variant is detected; laboratories may also want to consider reporting the results from the associated polyT tract in the partner of an individual who had a pathogenic or likely pathogenic variant detected during screening” (Deignan et al., 2020).
ACMG released recommendations for preconception and prenatal carrier screening. The following tier-based approach to carrier screening was produced and recommendations were made (Gregg et al., 2021):

CF: cystic fibrosis, SMA: spinal muscular atrophy.
- Preconception screening is recommended over prenatal screening.
- Concurrent partner testing should be offered if testing done during pregnancy.
- Tier 3 screening is recommended which includes 97 autosomal recessive genes and 16 X-linked genes, including DMD and Fragile X.
- Male partners may be offered Tier 3 carrier screening (for autosomal recessive conditions) if carrier screening is being performed at the same time with female partner.
- All pregnant patients and those planning a pregnancy should be offered Tier 3 carrier screening.
- Tier 4 screening should be considered when a pregnancy arises from a known or possible consanguineous relationship (second cousins or closer) or when a family or personal medical history warrants further risk assessment.
- ACMG does not recommend offering Tier 1 and/or Tier 2 screening, because these do not provide equitable evaluation of all racial/ethnic groups and routine offering of Tier 4 panels (Gregg et al., 2021).
National Society of Genetic Counselors (NSGC)
The National Society of Genetic Counselors (NSGC) recommends that “Carrier testing for CF should be offered to all women of reproductive age, regardless of ancestry; preferably pre-conceptionally. CF carrier testing should also be offered to any individual with a family history of CF and to partners of mutation carriers and people with CF” (Langfelder-Schwind et al., 2014).
Regarding what mutations should be included in the carrier screening test, the NSGC states, “Carrier testing panels should include the mutations recommended by ACOG and ACMG. For individuals of non- Northern European descent, pan-ethnic panels that include additional mutations more commonly identified in minority populations are appropriate to consider. Focus general population CF screening practices on identifying carriers of established disease-causing CFTR mutations” (Langfelder-Schwind et al., 2014).
The NSGC agrees with the ACMG regarding testing for IVS 8 5T/7T/9T as a reflex when mutation R117H is found in the CF carrier screen. They also assert that “in the absence of an R117H mutation, assessment of the intron 8 polyT or TG tracts is not recommended for routine CF carrier testing” (Langfelder-Schwind et al., 2014).
European Cystic Fibrosis Society (ECFS)
The ECFS mentioned CFTR modulators in their standards of care guideline. Recommendations state that “in patients with the G551D mutation ivacaftor should be part of standard of care”; at the time of writing, only one CFTR modulator had been shown to “demonstrate clinical efficacy” (Smyth et al., 2014).
In 2018, the ECFS published a revised version of their best practice guidelines. The ECFS has published the following requirement to when providing a CF diagnosis:
- “To be able to perform genetic testing for the most appropriate panel of CFTR mutations for the local population. Access to extended exon DNA analysis should be available when required” (Castellani et al., 2018).
The ECFS also states that genetic counseling should be offered when reporting a CF diagnosis to a patient.
National Institute for Health and Care Excellence (NICE)
In 2017, the NICE published guidelines on the diagnosis and management of CF. NICE includes the following recommendations on CF diagnoses:
- “Be aware that cystic fibrosis can be diagnosed based on:
- positive test results in people with no symptoms, for example infant screening (blood spot immunoreactive trypsin test) followed by sweat and gene tests for confirmation or
- clinical manifestations, supported by sweat or gene test results for confirmation or
- clinical manifestations alone, in the rare case of people with symptoms who have normal sweat or gene test results.
- Assess for cystic fibrosis and, when clinically appropriate, perform a sweat test (for children and young people) or a cystic fibrosis gene test (for adults) in people with any of the following:
- family history
- congenital intestinal atresia
- meconium ileus
- symptoms and signs that suggest distal intestinal obstruction syndrome
- faltering growth (in infants and young children)
- undernutrition
- recurrent and chronic pulmonary disease, such as:
- recurrent lower respiratory tract infections
- clinical or radiological evidence of lung disease (in particular bronchiectasis)
- persistent chest X-ray changes
- chronic wet or productive cough
- chronic sinus disease
- obstructive azoospermia (in young people and adults)
- acute or chronic pancreatitis
- malabsorption
- rectal prolapse (in children)
- pseudo-Bartter syndrome.
- Refer people with suspected cystic fibrosis to a specialist cystic fibrosis center if:
- they have a positive or equivocal sweat test result
- their assessment suggests they have cystic fibrosis but their test results are normal
- gene testing reveals 1 or more cystic fibrosis mutations” (NICE, 2017).
Royal College of Paediatrics and Child Health (RCPCH)
The RCPCH has published guidelines on the performance of the sweat test for the investigation of CF. These guidelines state that for patients less than six months of age and a sweat chloride level less than 30 mmol/L, “Cystic fibrosis is unlikely but requires genetic and clinical correlation”; also, for those older than six months and a sweat chloride level less than 40 mmol/L, “Cystic fibrosis is unlikely but requires genetic and clinical correlation” (RCPCH, 2014). Finally, the RCPCH notes that “The presence of two mutations of the CFTR gene known to cause CF may provide confirmatory evidence but the demonstration of mutations is not necessary to make a diagnosis of CF. The genetic diagnosis of CF by the demonstration of two CFTR mutations (known to be associated with clinical disease) does not require confirmation by a sweat test, however the UK Newborn Screening Program does currently require a sweat test after a positive newborn screening test, even in the presence of two CFTR mutations” (RCPCH, 2014).
Physicians Committee For Responsible Medicine (PCRM)
PSRM has published guidelines on diagnosis of cystic fibrosis. Below are the recommended guidelines:
- A sweat chloride test can be performed as early as 48 hours after birth, which is the mainstay of laboratory confirmation.
- Newborn Screening is mandatory in all 50 states.
A patient with symptoms may require a sweat chloride test or genetic testing. “Patients who exhibit signs and symptoms but do not have a positive sweat test should have a genetic test. Prenatal testing and newborn screening may be used. Early detection renders a better clinical course” (PCRM, 2021).
Table of Terminology
|
Term |
Definition |
|
ACMG |
American College of Medical Genetics |
|
ACOG |
American College of Obstetrics and Gynecology |
|
CAVD |
Congenital absence of the vas deferens |
|
CBAVD |
Congenital bilateral absence of the vas deferens |
|
CF |
Cystic fibrosis |
|
CFCD |
Cystic fibrosis carrier detection |
|
CFF |
Cystic Fibrosis Foundation |
|
CFSPID |
Cystic fibrosis screen positive inconclusive diagnosis |
|
CFTR |
Cystic fibrosis transmembrane conductance regulator |
|
CFTR2 |
Clinical and functional translation of CFTR |
|
CLIA ’88 |
Clinical Laboratory Improvement Amendments of 1988 |
|
CLSI |
Clinical and Laboratory Standards Institute |
|
CMS |
Centers for Medicare & Medicaid Services |
|
CPIC |
Clinical Pharmacogenetics Implementation Consortium |
|
CRMS |
Customer relationship management system |
|
CVS |
Chorionic villus sampling |
|
DNA |
Deoxyribonucleic acid |
|
ECFS |
European Cystic Fibrosis Society |
|
FDA |
Food and Drug Administration |
|
FEV1 |
Forced expiratory volume in 1 second |
|
ICM |
Intestinal current measurement |
|
IRT |
Immunoreactive trypsinogen |
|
IVA |
Ivacaftor |
|
LDT |
Laboratory-developed test |
|
LUM |
Lumacaftor |
|
MMWR |
Morbidity And Mortality Weekly Report |
|
MVCC |
Mutation of varying clinical consequence |
|
NBS |
Newborn screening |
|
NGS |
Next generation sequencing |
|
NICE |
National Institute for Health and Care Excellence |
|
NPD |
Nasal potential difference |
|
NSGC |
National Society of Genetic Counselors |
|
PCR |
Polymerase chain reaction |
|
PCRM |
Physicians Committee for Responsible Medicine |
|
PCS |
Cystic fibrosis carrier screening |
|
QALY |
Quality adjusted life year |
|
RCPCH |
Royal College of Paediatrics and Child Health |
|
RT-PCR |
Reverse transcription polymerase chain reaction |
|
TDAS |
XTAG Data Analysis Software |
|
TDN |
Therapeutics Development Network |
|
TOP |
Termination of pregnancy |
|
UNK |
Uncharacterized mutation |
|
VUS |
Variant of uncertain significance |
Reference
- ACMG. (2011). Technical Standards and Guidelines for CFTR Mutation Testing. STANDARDS AND GUIDELINES FOR CLINICAL GENETICS LABORATORIES. http://dx.doi.org/10.1038/gim.2014.93
- ACOG. (2017). Committee Opinion No. 691: Carrier Screening for Genetic Conditions. Obstet Gynecol, 129(3), e41-e55. https://doi.org/10.1097/aog.0000000000001952
- Avram, C. M., Dyer, A. L., Shaffer, B. L., & Caughey, A. B. (2021). The cost-effectiveness of genotyping versus sequencing for prenatal cystic fibrosis carrier screening. Prenatal Diagnosis, 41(11), 1449-1459. https://doi.org/https://doi.org/10.1002/pd.6027
- Baker, M. W., Atkins, A. E., Cordovado, S. K., Hendrix, M., Earley, M. C., & Farrell, P. M. (2016). Improving newborn screening for cystic fibrosis using next-generation sequencing technology: a technical feasibility study. Genet Med, 18(3), 231-238. https://doi.org/10.1038/gim.2014.209
- Barrett, P. M., Alagely, A., & Topol, E. J. (2012). Cystic fibrosis in an era of genomically guided therapy. Hum Mol Genet, 21(R1), R66-71. https://doi.org/10.1093/hmg/dds345
- Bell, S. C., De Boeck, K., & Amaral, M. D. (2015). New pharmacological approaches for cystic fibrosis: promises, progress, pitfalls. Pharmacol Ther, 145, 19-34. https://doi.org/10.1016/j.pharmthera.2014.06.005
- Castellani, C., Duff, A. J. A., Bell, S. C., Heijerman, H. G. M., Munck, A., Ratjen, F., Sermet-Gaudelus, I., Southern, K. W., Barben, J., Flume, P. A., Hodkova, P., Kashirskaya, N., Kirszenbaum, M. N., Madge, S., Oxley, H., Plant, B., Schwarzenberg, S. J., Smyth, A. R., Taccetti, G., . . . Drevinek, P. (2018). ECFS best practice guidelines: the 2018 revision. J Cyst Fibros, 17(2), 153-178. https://doi.org/10.1016/j.jcf.2018.02.006
- Celera. (2007). Cystic Fibrosis Genotyping Assay. https://www.accessdata.fda.gov/cdrh_docs/pdf6/K062028.pdf
- CF Foundation, U., Johns Hopkins University, The Hospital for Sick Children. (2021). The Clinical and Functional TRanslation of CFTR (CFTR2). https://www.cftr2.org/resources
- Clancy, J. P., & Jain, M. (2012). Personalized medicine in cystic fibrosis: dawning of a new era. Am J Respir Crit Care Med, 186(7), 593-597. https://doi.org/10.1164/rccm.201204-0785PP
- Clancy, J. P., Johnson, S. G., Yee, S. W., McDonagh, E. M., Caudle, K. E., Klein, T. E., Cannavo, M., & Giacomini, K. M. (2014). Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for ivacaftor therapy in the context of CFTR genotype. Clin Pharmacol Ther, 95(6), 592-597. https://doi.org/10.1038/clpt.2014.54
- Clinical_Micro_Sensors. (2006). eSensor® Cystic Fibrosis Carrier Detection System. https://pathsurveyor.com/api/pmn_pdf?knumber=K060543#:~:text=The%20eSensor%C2%AE%20Cystic%20Fibrosis%20Carrier%20Detection%20System%20is%20an,isolated%20from%20human%20whole%20blood.
- Cohen-Cymberknoh, M., Shoseyov, D., & Kerem, E. (2011). Managing cystic fibrosis: strategies that increase life expectancy and improve quality of life. Am J Respir Crit Care Med, 183(11), 1463-1471. https://doi.org/10.1164/rccm.201009-1478CI
- Deignan, J. L., Astbury, C., Cutting, G. R., del Gaudio, D., Gregg, A. R., Grody, W. W., Monaghan, K. G., Richards, S., & Committee, A. L. Q. A. (2020). CFTR variant testing: a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genetics In Medicine, 22(8), 1288-1295. https://doi.org/10.1038/s41436-020-0822-5
- Farrell, P. M., Rosenstein, B. J., White, T. B., Accurso, F. J., Castellani, C., Cutting, G. R., Durie, P. R., Legrys, V. A., Massie, J., Parad, R. B., Rock, M. J., & Campbell, P. W., 3rd. (2008). Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. J Pediatr, 153(2), S4-s14. https://doi.org/10.1016/j.jpeds.2008.05.005
- Farrell, P. M., White, T. B., Ren, C. L., Hempstead, S. E., Accurso, F., Derichs, N., Howenstine, M., McColley, S. A., Rock, M., Rosenfeld, M., Sermet-Gaudelus, I., Southern, K. W., Marshall, B. C., & Sosnay, P. R. (2017). Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J Pediatr, 181s, S4-S15.e11. https://doi.org/10.1016/j.jpeds.2016.09.064
- FDA. (2006a). Decision Summary ESENSOR CYSTIC FIBROSIS CARRIER DETECTION TEST 510(k) Premarket Notification. http://www.accessdata.fda.gov/cdrh_docs/reviews/K051435.pdf
- FDA. (2006b). Decision Summary Tag It Cystic Fibrosis Carrier Detection System 510(k) Premarket Notification. Retrieved from https://www.accessdata.fda.gov/cdrh_docs/reviews/K043011.pdf
- FDA. (2007). Decision Summary Cystic Fibrosis Genotyping Assay 510(k) Premarket Notification. U.S. Food and Drug Administration Retrieved from https://www.accessdata.fda.gov/cdrh_docs/reviews/K062028.pdf
- FDA. (2008). Decision Summary Inplex CF Molecular Test 510(k) Premarket Notification. U.S. Food and Drug Administration Retrieved from https://www.accessdata.fda.gov/cdrh_docs/reviews/K063787.pdf
- FDA. (2009a). Decision Summary eSensor® CF Genotyping Test 510(k) Premarket Notification. U.S. Food and Drug Administration Retrieved from https://www.accessdata.fda.gov/cdrh_docs/reviews/K090901.pdf
- FDA. (2009b). Decision Summary Verigene® CFTR and Verigene® CFTR PolyT Nucleic Acid Tests 510(k) Premarket Notification. U.S. Food and Drug Administration Retrieved from https://www.accessdata.fda.gov/cdrh_docs/reviews/K083294.pdf
- FDA. (2009c). Decision Summary xTAG® Cystic Fibrosis 60 Kit v2 510(k) Premarket Notification. U.S. Food and Drug Administration Retrieved from https://www.accessdata.fda.gov/cdrh_docs/reviews/K083845.pdf
- FDA. (2013a). Decision Summary Illumina MiSeqDxTM Cystic Fibrosis Clinical Sequencing Assay 510(k) Premarket Notification. U.S. Food and Drug Administration Retrieved from https://www.accessdata.fda.gov/cdrh_docs/reviews/K132750.pdf
- FDA. (2013b). Decision Summary Illumina MiSeqDx™ Cystic Fibrosis 139-Variant Assay 510(k) Premarket Notification. U.S. Food and Drug Administration Retrieved from https://www.accessdata.fda.gov/cdrh_docs/reviews/K124006.pdf
- FDA. (2016a). XTAG Cystic Fibrosis 39 Kit V2. https://www.accessdata.fda.gov/scripts/cdrh/devicesatfda/index.cfm?start_search=1&q=Y3lzdGljIGZpYnJvc2lz&approval_date_from=&approval_date_to=&sort=approvaldatedesc&pagenum=10
- FDA. (2016b). XTAG Cystic Fibrosis 60 Kit V2, XTAG Data Analysis Software (TDAS) CFTR. https://www.accessdata.fda.gov/scripts/cdrh/devicesatfda/index.cfm?db=pmn&id=K163336
- Gregg, A. R., Aarabi, M., Klugman, S., Leach, N. T., Bashford, M. T., Goldwaser, T., Chen, E., Sparks, T. N., Reddi, H. V., Rajkovic, A., Dungan, J. S., Practice, A. P., & Guidelines, C. (2021). Screening for autosomal recessive and X-linked conditions during pregnancy and preconception: a practice resource of the American College of Medical Genetics and Genomics (ACMG). Genetics In Medicine, 23(10), 1793-1806. https://doi.org/10.1038/s41436-021-01203-z
- Grosse, S. D., Boyle, C. A., Botkin, J. R., Comeau, A. M., Kharrazi, M., Rosenfeld, M., & Wilfond, B. S. (2004). Newborn screening for cystic fibrosis: evaluation of benefits and risks and recommendations for state newborn screening programs. MMWR Recomm Rep, 53(Rr-13), 1-36. http://dx.doi.org/
- Hendrix, M. M., Cuthbert, C. D., & Cordovado, S. K. (2020). Assessing the Performance of Dried-Blood-Spot DNA Extraction Methods in Next Generation Sequencing. International Journal of Neonatal Screening, 6(2), 36. https://www.mdpi.com/2409-515X/6/2/36
- Hologic. (2008). InPlex Molecular Test. http://www.hologic.ca/sites/default/files/package%20inserts/Hologic%20CF%20IVD%20Package%20Insert.pdf
- Illumina. (2013). Illumina MiSeqDx™ Cystic Fibrosis 139-Variant Assay. https://support.illumina.com/content/dam/illumina-marketing/documents/clinical/miseqdx-cystic-fibrosis-139-variant-assay-data-sheet-1000000006359.pdf
- Kessels, S. J. M., Carter, D., Ellery, B., Newton, S., & Merlin, T. L. (2020). Prenatal genetic testing for cystic fibrosis: a systematic review of clinical effectiveness and an ethics review. Genet Med, 22(2), 258-267. https://doi.org/10.1038/s41436-019-0641-8
- Langfelder-Schwind, E., Karczeski, B., Strecker, M. N., Redman, J., Sugarman, E. A., Zaleski, C., Brown, T., Keiles, S., Powers, A., Ghate, S., & Darrah, R. (2014). Molecular testing for cystic fibrosis carrier status practice guidelines: recommendations of the National Society of Genetic Counselors. J Genet Couns, 23(1), 5-15. https://doi.org/10.1007/s10897-013-9636-9
- Lim, R. M., Silver, A. J., Silver, M. J., Borroto, C., Spurrier, B., Petrossian, T. C., Larson, J. L., & Silver, L. M. (2016). Targeted mutation screening panels expose systematic population bias in detection of cystic fibrosis risk. Genet Med, 18(2), 174-179. https://doi.org/10.1038/gim.2015.52
- Luminex. (2016). xTAG® Cystic Fibrosis 71 v2 Kit https://www.luminexcorp.com/cystic-fibrosis/#overview
- Lyon, E., Schrijver, I., Weck, K. E., Ferreira-Gonzalez, A., Richards, C. S., & Palomaki, G. E. (2014). Molecular genetic testing for cystic fibrosis: laboratory performance on the College of American Pathologists external proficiency surveys [Original Research Article]. Genetics In Medicine, 17, 219. https://doi.org/10.1038/gim.2014.93
- Morris-Rosendahl, D. J., Edwards, M., McDonnell, M. J., John, S., Alton, E. W., Davies, J. C., & Simmonds, N. J. (2020). Whole Gene Sequencing of CFTR Reveals a High Prevalence of the Intronic Variant c.3874-4522A>G in Cystic Fibrosis. Am J Respir Crit Care Med. https://doi.org/10.1164/rccm.201908-1541LE
- Nanosphere. (2009). Verigene® CFTR and Verigene® CFTR PolyT Nucleic Acid Tests. https://www.accessdata.fda.gov/cdrh_docs/reviews/K083294.pdf
- NICE. (2017). Cystic fibrosis: diagnosis and management. https://www.nice.org.uk/guidance/ng78/chapter/Recommendations#diagnosis-of-cystic-fibrosis
- PCRM. (2021). Cystic Fibrosis. https://nutritionguide.pcrm.org/nutritionguide/view/Nutrition_Guide_for_Clinicians/1342064/all/Cystic_Fibrosis
- Pletcher, B. A., & Bocian, M. (2006). Preconception and prenatal testing of biologic fathers for carrier status. American College of Medical Genetics. Genet Med, 8(2), 134-135. https://doi.org/10.109701.gim.0000200948.58427.e2
- Pritchard, L. L. (2016). Respiratory Conditions Update: Cystic Fibrosis. FP Essent, 448, 35-43. https://www.ncbi.nlm.nih.gov/pubmed/27576234
- RCPCH. (2014). Guidelines for the Performance of the Sweat Test for the Investigation of Cystic Fibrosis in the UK 2nd Version http://www.acb.org.uk/docs/default-source/committees/scientific/guidelines/acb/sweat-guideline-v2-1.pdf
- Ren, C. L., Morgan, R. L., Oermann, C., Resnick, H. E., Brady, C., Campbell, A., DeNagel, R., Guill, M., Hoag, J., Lipton, A., Newton, T., Peters, S., Willey-Courand, D. B., & Naureckas, E. T. (2018). Cystic Fibrosis Foundation Pulmonary Guidelines. Use of Cystic Fibrosis Transmembrane Conductance Regulator Modulator Therapy in Patients with Cystic Fibrosis. Ann Am Thorac Soc, 15(3), 271-280. https://doi.org/10.1513/AnnalsATS.201707-539OT
- Schrijver, I., Pique, L., Graham, S., Pearl, M., Cherry, A., & Kharrazi, M. (2016). The Spectrum of CFTR Variants in Nonwhite Cystic Fibrosis Patients: Implications for Molecular Diagnostic Testing. J Mol Diagn, 18(1), 39-50. https://doi.org/10.1016/j.jmoldx.2015.07.005
- Sharma, D., Xing, S., Hung, Y. T., Caskey, R. N., Dowell, M. L., & Touchette, D. R. (2018). Cost-effectiveness analysis of lumacaftor and ivacaftor combination for the treatment of patients with cystic fibrosis in the United States. Orphanet J Rare Dis, 13(1), 172. https://doi.org/10.1186/s13023-018-0914-3
- Sicko, R. J., Stevens, C. F., Hughes, E. E., Leisner, M., Ling, H., Saavedra-Matiz, C. A., Caggana, M., & Kay, D. M. (2021). Validation of a Custom Next-Generation Sequencing Assay for Cystic Fibrosis Newborn Screening. Int J Neonatal Screen, 7(4). https://doi.org/10.3390/ijns7040073
- Smyth, A. R., Bell, S. C., Bojcin, S., Bryon, M., Duff, A., Flume, P., Kashirskaya, N., Munck, A., Ratjen, F., Schwarzenberg, S. J., Sermet-Gaudelus, I., Southern, K. W., Taccetti, G., Ullrich, G., & Wolfe, S. (2014). European Cystic Fibrosis Society Standards of Care: Best Practice guidelines. Journal of Cystic Fibrosis, 13, S23-S42. https://doi.org/10.1016/j.jcf.2014.03.010
- Sosnay, P. R., Siklosi, K. R., Van Goor, F., Kaniecki, K., Yu, H., Sharma, N., Ramalho, A. S., Amaral, M. D., Dorfman, R., Zielenski, J., Masica, D. L., Karchin, R., Millen, L., Thomas, P. J., Patrinos, G. P., Corey, M., Lewis, M. H., Rommens, J. M., Castellani, C., . . . Cutting, G. R. (2013). Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat Genet, 45(10), 1160-1167. https://doi.org/10.1038/ng.2745
- Stafler, P., Mei-Zahav, M., Wilschanski, M., Mussaffi, H., Efrati, O., Lavie, M., Shoseyov, D., Cohen-Cymberknoh, M., Gur, M., Bentur, L., Livnat, G., Aviram, M., Alkrinawi, S., Picard, E., Prais, D., Steuer, G., Inbar, O., Kerem, E., & Blau, H. (2016). The impact of a national population carrier screening program on cystic fibrosis birth rate and age at diagnosis: Implications for newborn screening. J Cyst Fibros, 15(4), 460-466. https://doi.org/10.1016/j.jcf.2015.08.007
- Straniero, L., Solda, G., Costantino, L., Seia, M., Melotti, P., Colombo, C., Asselta, R., & Duga, S. (2016). Whole-gene CFTR sequencing combined with digital RT-PCR improves genetic diagnosis of cystic fibrosis. J Hum Genet. https://doi.org/10.1038/jhg.2016.101
- Sugunaraj, J. P., Brosius, H. M., Murray, M. F., Manickam, K., Stamm, J. A., Carey, D. J., & Mirshahi, U. L. (2019). Predictive value of genomic screening: cross-sectional study of cystic fibrosis in 50,788 electronic health records. NPJ Genom Med, 4, 21. https://doi.org/10.1038/s41525-019-0095-6
- Tm_Bioscience_Corporation. (2006). Tag-It™ Cystic Fibrosis Kit. https://www.accessdata.fda.gov/cdrh_docs/reviews/K043011.pdf
- Wainwright, C. E., Elborn, J. S., Ramsey, B. W., Marigowda, G., Huang, X., Cipolli, M., Colombo, C., Davies, J. C., De Boeck, K., Flume, P. A., Konstan, M. W., McColley, S. A., McCoy, K., McKone, E. F., Munck, A., Ratjen, F., Rowe, S. M., Waltz, D., & Boyle, M. P. (2015). Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med, 373(3), 220-231. https://doi.org/10.1056/NEJMoa1409547
- Watson, M. S., Cutting, G. R., Desnick, R. J., Driscoll, D. A., Klinger, K., Mennuti, M., Palomaki, G. E., Popovich, B. W., Pratt, V. M., Rohlfs, E. M., Strom, C. M., Richards, C. S., Witt, D. R., & Grody, W. W. (2004). Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genet Med, 6(5), 387-391. https://doi.org/10.109701.gim.0000139506.11694.7c
- Wilschanski, M., Zielenski, J., Markiewicz, D., Tsui, L. C., Corey, M., Levison, H., & Durie, P. R. (1995). Correlation of sweat chloride concentration with classes of the cystic fibrosis transmembrane conductance regulator gene mutations. J Pediatr, 127(5), 705-710. http://dx.doi.org/
Coding Section
| Code | Number | Description |
| CPT | 81220 | CFTR (cystic fibrosis transmembrane conductance regulator) (e.g., cystic fibrosis) gene analysis; common variants (e.g., ACMG/ACOG guidelines). (When Intron 8 poly-T analysis is performed in conjunction with 81220 in a R117H positive patient, do not report 81224) |
| 81221 | Known familial variants | |
| 81222 | Duplication/deletion variants | |
| 81223 | Full gene sequence | |
| 81224 | CFTR gene intron 8 poly-T analysis (e.g., male infertility) | |
| 81412 | Ashkenazi Jewish associated disorders (e.g., Bloom syndrome, Canavan disease, cystic fibrosis, familial dysautonomia, Fanconi anemia group C, Gaucher disease, Tay-Sachs disease), genomic sequence analysis panel, must include sequencing of at least 9 genes, including ASPA, BLM, CFTR, FANCC, GBA, HEXA, IKBKAP, MCOLN1, and SMPD1 | |
| 81479 | Unlisted molecular pathology code |
Procedure and diagnosis codes on Medical Policy documents are included only as a general reference tool for each policy. They may not be all-inclusive.
This medical policy was developed through consideration of peer-reviewed medical literature generally recognized by the relevant medical community, U.S. FDA approval status, nationally accepted standards of medical practice and accepted standards of medical practice in this community, Blue Cross Blue Shield Association technology assessment program (TEC) and other nonaffiliated technology evaluation centers, reference to federal regulations, other plan medical policies, and accredited national guidelines.
"Current Procedural Terminology © American Medical Association. All Rights Reserved"
History From 2017 Forward
| 08/10/2022 | Annual review, policy reformatted for clarity, adding coverage criteria #7. Also updating descrition, rationale and references. |
|
07/29/2021 |
Annual review, updating policy for specificity of #3 adn #4. No change to policy intent. |
|
07/22/2020 |
Annual review, no change to policy intent. Updating background, guidelines and references. |
|
07/12/2019 |
Annual review, no change to policy intent. |
|
06/19/2019 |
Interim review. Genetic counseling is recommended is replacing Genetic counseling is Medically necessary. No other changes made. |
|
07/18/2018 |
Annual review, no change to policy intent. |
|
10/09/2017 |
Updated Gene Testing for Cystic Fibrosis table. No other changes |
|
07/19/2017 |
New Policy |