
Genetic Testing for Fanconi Anemia - CAM 345
Description
Fanconi anemia (FA) is an inherited disorder in which cells cannot correctly repair inter-strand crosslinks (ICLs), a specific type of DNA damage that results in genomic instability. This can lead to bone marrow failure (such as aplastic anemia), leukemia, and/or solid tumors. FA is rare, occurring in 1 in 100,000 to 250,000 births, with an increased incidence in populations such as Ashkenazi Jews and South African Afrikaner populations (Olson, 2022).
Regulatory Status
No U.S. Food and Drug Administration-cleared genetic tests for FA were found as of 09/23/2020. Thus, the tests are offered as laboratory-developed tests. Many labs have developed specific tests that they must validate and perform in house. These laboratory-developed tests (LDTs) are regulated by the Centers for Medicare & Medicaid (CMS) as high-complexity tests under the Clinical Laboratory Improvement Amendments of 1988 (CLIA ’88). As an LDT, the U.S. Food and Drug Administration has not approved or cleared this test; however, FDA clearance or approval is not currently required for clinical use.
Policy
- For individuals who have received genetic counseling and who have clinical signs and symptoms of Fanconi anemia (FA), genetic testing for the diagnosis of FA is considered MEDICALLY NECESSARY.
- For pregnant individuals and those seeking pre-conceptive care, carrier screening for FA is considered MEDICALLY NECESSARY.
- In situations where both biological parents are known carriers of a pathogenic FA mutation or where one biological parent is FA-affected and the other biological parent. is a known carrier of a pathogenic FA mutation, preimplantation genetic testing for FA is considered MEDICALLY NECESSARY.
- For all other situations not discussed above, genetic testing for the diagnosis of FA is considered NOT MEDICALLY NECESSARY.
Rationale
Primarily inherited as an autosomal recessive disorder, Fanconi anemia (FA) is associated with known mutations in at least 22 FA identified genes (Jung et al., 2020). It is found equally in males and females, as well as in different ethnic groups; approximately 50% of FA patients are diagnosed by age 10 (NORD, 2020). Jung et al. (2020) also noted that siblings with FA often have similar hematological courses, potentially attributed to similarity in causative variants and environmental factors but have different presentations of congenital anomalies (except for kidney abnormalities and microcephaly to a moderate degree). The three most commonly mutated genes in FA are FANCA, FANCC, and FANCG; these comprise up to 80-90% of all FA cases, with FANCA mutations accounting for approximately 60% of cases worldwide (Bogliolo et al., 2019; Olson, 2022). The main function of this set of proteins is to repair the inter-strand crosslinks (ICL) that typically form during DNA replication and transcription (Olson, 2022). A cell is estimated to repair about 10 ICLs per day, but as few as 20-40 unresolved ICLs can lead to cell death (Sumpter & Levine, 2017). The FA pathway may also play a role in other functions, such as metabolizing alcohol, ensuring the stability of the replication fork during DNA replication, managing oxidative stress as in providing defense from reactive oxygen species (ROS)-induced cell death, and repairing double strand breaks (Kottemann & Smogorzewska, 2013; Longerich et al., 2014; Milletti et al., 2020; Olson, 2022). For example, a mutation in the FANCC gene was found to impede the cell’s ability to clear out damaged mitochondria and viruses, which could eventually lower immunity to viral infection and contribute to the characteristic bone marrow failure (Cheung & Taniguchi, 2017; Sumpter et al., 2016).
FA may manifest in several ways with symptoms including short stature, skin findings such as hyper- or hypo- pigmentation and café-au-lait skin lesions, microcephaly, and abnormalities in the thumb, eye, axial skeleton, ear, renal system, or urinary tract. There is also a potential connection between FA and the VACTERL-H association (three or more of the following: vertebral anomalies, anal atresia, congenital heart disease, tracheoesophageal fistula, esophageal atresia, renal anomalies, limb anomalies, and hydrocephalus) as the percentage of FA patients also meeting the criteria for VACTERL-H was much higher than previously found (Alter & Giri, 2016). However, up to 25 – 40% of FA patients look physically normal (D'Andrea, 2010). At the physiological level, the most common symptoms are bone marrow failure and cytopenias, such as pancytopenia, macrocytic anemia, or thrombocytopenia (Olson, 2022). Bone marrow failure is reportedly the most common primary symptom in FA and presents itself in 70 – 80% of patients by age ten (Bogliolo et al., 2019). Though the exact mechanism of premature hematopoietic stem cell (HSC) loss in FA remains unclear, it is thought to be impacted by defective DNA repair, causing increased damage and cell cycle arrest, increased levels of reactive oxygen species and inflammatory cytokines, and damage caused by reactive aldehydes in the absence of intact repair pathways (Olson, 2022). Aplastic anemia, another common FA side effect which causes the body to halt the production of red blood cells, also typically occurs early, either leading to death or to a hematopoietic stem cell transplant. Endocrine issues, such as growth hormone deficiency, abnormal glucose/insulin metabolism, dyslipidemia, pubertal delay, and hypothyroidism are also commonly associated with an FA diagnosis (about 80% of FA children and adults have at least one endocrine defect) and often lead to a worsening life quality in FA patients (Milletti et al., 2020; Shimamura & Alter, 2010).
Screening for Fanconi Anemia
The most common screening assay for Fanconi anemia is the chromosome breakage test. A DNA cross-linking agent, such as mitomycin C (MMC) or diepoxybutane (DEB), is used to induce chromosome breakage, and the cells are evaluated at their respective stages in the cell cycle. FA cells typically have more DNA damage and are forced to arrest in the G2 phase when these cells can be observed. Tests may be positive, negative, or inconclusive; a positive test typically shows about 90% of lymphocytes with increased breakage, a negative test shows no increased breakage, and an inconclusive test cannot provide any definitive answer (Hays, 2014). Normal cells have a mean baseline of < .05 breaks per cell while FA cells may range from 0.02 – 0.85 breaks per cell. DEB (the more sensitive and specific agent) typically has a mean baseline of < .10 breaks per normal cell and from 1.06 to 23.9 breaks per FA cell (Auerbach, 2015). The International Fanconi Anemia Registry (IFAR) found the mean standard error of breaks per cell of 104 FA patients to be 8.96 ± 0.448 and the mean standard error of percentage of cells with breaks to be 85.15% ± 1.99%, compared to 0.06 ± 0.004 breaks and 5.12% ± 0.28% of 224 non-FA patients (Kook et al., 1998).
Inconclusive results are typically due to one of two possibilities — one is “mosaicism,” where two separate populations of lymphocytes in the blood occur, and the other is where the patient has another underlying condition causing chromosome breakage. However, a mutation analysis can corroborate a diagnosis or provide further information. This can be particularly helpful in assessing the patient’s family members, such as potential carriers, asymptomatic family members, or members who may develop clinical symptoms (FARF, 2020).
More recently, researchers have utilized whole exome sequencing as a diagnostic method for FA. Historically, molecular diagnostics regarding FA have been challenging for the medical community because the disease is caused by hereditary patterns featuring point mutations and large genomic deletions in an estimated 22 genes (Rio et al., 2019). Nonetheless, the whole exome sequencing method used in this study identified 93.3% of deletions and mutated alleles when compared to a previously validated method, leading the researchers to the conclusion that whole exome sequencing is efficient enough to characterize patients with FA (Rio et al., 2019).
Clinical Utility and Validity
Due to the increased instability of an FA patient’s genome, it is common to see an increased risk of cancer in patients with FA, particularly bone marrow cancers such as leukemia. A study found the observed to expected ratio of all cancers to be as high as 48 (i.e. the observed rate was 48 times higher than expected after controlling for factors such as age and sex) (Alter, 2014). The same study found the likelihood that an FA patient would develop acute myeloid leukemia (AML) to be 700 times higher than normal and the likelihood to develop any myelodysplastic syndrome (MDS) to be 6000 times higher (Alter, 2014). Underlying FA disease mechanisms may also be causing patients to develop cancers at a much earlier age than typically observed. A study focusing on 35 FA patients found that when compared to the general population, those afflicted by FA were, on average, diagnosed with head and neck squamous cell carcinoma 31 years earlier than controls (32 years for FA patients, 63 years for general population). FA mutation type may also play a factor in cancer development as another study found that FA patients with FANCA mutations developed cancer at a significantly older age than those with other mutations; however, mutation type did not seem to affect the overall survival rates of FA cancer patients (Steinberg-Shemer et al., 2019). Furthermore, the common risk factors, such as tobacco or alcohol consumption, were typically not a factor for the FA patients as is usually seen in the general population (Kutler et al., 2016).
Another example of how intertwined the FA proteins and pathway is with cancer is found in the FANCD1 (Fanconi anemia complementation group D1) gene. The FANCD1 gene, also known as BRCA2, is a gene whose mutations often lead to a higher risk of breast cancer. The BRCA2 (-/-) cell reacts the same way an FA cell does when treated with the crosslinking agents and BRCA2 co-localizes with FANCD2 at damaged sections of DNA. The patients with heterozygous genotypes of BRCA2 are historically more likely to have a higher risk of breast and ovarian cancer (D'Andrea, 2010).
Novel studies have further demonstrated the risk of germline mutations in FA complementation group (FANC) of the FA pathway in cancer. FANCD2 (Fanconi anemia complementation group D2) was found to confer a malignant phenotype in esophageal squamous cell carcinoma, and cyclin-cyclin-dependent kinase (CDK) and ataxia-telangiectasia RAD3-related/ataxia-telangiectasia mutated (ATR/ATM) signaling was shown to help in depletion of FANCD2 protein expression and suppress cancer cell proliferation (Lei et al., 2020). In a different study done on a Han Chinese population, Yu et al. (2020) identified that Fanconi anemia complement group F (FANCF), though already known to impact cell proliferation and DNA repair, can increase risk of colorectal cancer if hypomethylated. Aberrant methylation of FANCF was also observed in ovarian tumors, non-small-cell lung cancer, cervical cancer, and oral cancer previously in general populations (Yu et al., 2020). This conveys the markedly increased predisposition to cancer via mutations in FA and FA pathway components.
Chang et al. (2021) discusses novel diagnostic approaches for FA by single-cell sequencing and capillary nano-immunoassay. Next-generation sequencing (NGS) has been widely utilized for FA diagnosis but has limitations that may lead to unconfirmed genetic subtypes. Chang et al. (2021) studies one FA cause with FANCM mutation by conducting the capillary-nano-immunoassay to analyze the expression profile of FA-associated proteins. This assay is designed to detect 417 blood disease genes, including the 17 known FA-related genes. In this case, Chang et al. (2021) observed two homozygous mutations of the FANCM and FANCD1 genes and abnormal expression of both genes simultaneously existed, diagnosing the patient as a FA-D1/FA-M dual subtype. According to the author, "compared with mixed cell sequencing, single-cell sequencing data shows more accuracy for the FA subtype evaluation, while the capillary nano-immunoassay is a good method to detect the expression profile of abnormal or modified FA protein (Chang et al., 2021).”
Chan et al. (2021) studied the genetic spectrum of FA-associated genes across populations of varying ancestries to explore potential genotype-phenotype associations in cancer. 3,523 subjects in Singapore, of varying ancestry, were assessed for carrier frequency and variant spectrum of potentially pathogenic variants in 17 FA genes. The data suggested higher germline and somatic mutation burden between FANCA and FANCC with head and neck and lung squamous cell carcinomas and FANCI and SLX4/FANCP with uterine cancer. Additionally, Chan et al. (2021) highlighted the differences in carrier frequencies in non-European populations, considering the fact that our knowledge about the clinical and genetic spectrum of FA is derived predominantly from populations of European ancestry. “Given the variable distribution of germline pathogenic variant carriers across different ancestries, genetic testing for molecular diagnosis should not be restricted to FANCA, FANCC, and FANCG, which are reported more frequently in FA patients of European descent, but should include all known FA genes.”
Alter et al. (2022) classified patients by age of diagnosis as part of a National Cancer Institute IBMFS cohort that included 178 pediatric patients and 26 adult patients. The authors were investigating whether FA cases could be distinguished and placed into subgroups by age diagnosed. The various features that were compared included the cumulative incidences of first adverse events (severe BMF leading to hematopoietic cell transplant or death, leukemia, or solid tumors) between an adult cohort and the pediatric cohort. The adult group did not consistently have the traditional FA features of birth defects, early-onset bone marrow failure or leukemia, and the group was mostly female with more patients with the FANCA genotype. This adult group developed more head and neck squamous-cell carcinoma and/or gynecological cancers (as compared to the pediatric group). The overall conclusion is Hematology and Oncology providers should investigate an FA diagnosis in adult patients that present with early-onset head and neck squamous-cell carcinoma or gynecological cancer (with or without hematologic issues) (Alter et al., 2022).
American College of Medical Genetics and Genomics
The guidelines for clinical genetics laboratories are specified in the 2018 (revised January 2021) edition of the Standards and Guidelines for Clinical Genetics Laboratories by the ACMG. The guidelines on FA state:
- A cytogenetic evaluation for FA should include an induction of breakage with a crosslinking agent such as MMC or DEB (in addition to a baseline chromosome breakage).
- A well-established negative and positive control should be present and multiple cultures are recommended (if there is enough specimen available).
- At least 50 different cells (banded or unbanded) in the metaphase stage of the cell cycle should be analyzed, and the percentage of cells with aberration should reported (ACMG, 2021)
In 2021, ACMG released an updated guideline for screening for autosomal recessive and X-linked conditions during pregnancy and preconception. Their practice resource reviews aim to recommend “a consistent and equitable approach for offering carrier screening to all individuals during pregnancy and preconception” and replaces any earlier ACMG position statements on prenatal/preconception expanded carrier screening and provide the following recommendations:
- "Analytical validity of carrier screening is to be established by a laboratory in compliance with CLIA/CAP regulations and adhering to ACMG Laboratory Standards and Guidelines.”
- As evidence evolves, ClinVar and ClinGen continually update pathogenicity of variants and the association between genes and conditions, respectively.”
- “Carrier screening enables those screened to consider their reproductive risks, reproductive options, and to make informed decisions.”
- “Published evidence supports clinical utility for carrier screening of multiple conditions simultaneously.”
- “The phrase ‘expanded carrier screening’ be replaced by ‘carrier screening.’”
- “Adopting a more precise tiered system based on carrier frequency:
- Tier 4: < 1/200 carrier frequency (includes Tier 3) genes/condition will vary by lab
- Tier 3: ≥ 1/200 carrier frequency (includes Tier 2) includes X-linked conditions
- Tier 2: ≥ 1/100 carrier frequency (includes Tier 1)
- Tier 1: CF [Cystic Fibrosis] + SMA [spinal muscular atrophy] + Risk Based Screening
Fanconi anemia falls into Tier 2 carrier screening, according to the ACMG recommendation. For purposes of Fanconi anemia screening, this policy focuses on Tier 2 and Tier 3 carrier screening (as Tier 3 is inclusive of Tier 2). ACMG recommends:
- “Tier 2 carrier screening stems from an ACOG recommendation for conditions that have a severe or moderate phenotype and a carrier frequency of at least 1/100.” However, “data demonstrate that carrier screening for two common conditions using a carrier frequency threshold of 1/100 may not be equitable across diverse populations. Others have shown that limiting the carrier frequency to ≥ 1/100 creates missed opportunities to identify couples at risk for serious conditions.”
- “We define Tier 3 screening as carrier screening for conditions with a carrier frequency ≥ 1/200 . . . Tier 2 and Tier 3 screening prioritize carrier frequency as a way to think about conditions most appropriate for screening in the general population. However, when ACOG proposed this level, they did not specify whether it was thinking about carrier frequency in terms of the global population or subpopulations. We use “carrier frequency” to mean in any ethnic group with reasonable representation in the United States.”
- “All pregnant patients and those planning a pregnancy should be offered Tier 3 carrier screening.”
- ACMG does NOT recommend:
- Offering Tier 1 and/or Tier 2 screening, because these do not provide equitable evaluation of all racial/ethnic groups.
- Routine offering of Tier 4 panels.
- “Carrier screening paradigms should be ethnic and population neutral and more inclusive of diverse populations to promote equity and inclusion.”
- “All pregnant patients and those planning a pregnancy should be offered Tier 3 carrier screening for autosomal recessive (Tables 1–5) and X-linked (Table 6) conditions.”
- “Reproductive partners of pregnant patients and those planning a pregnancy may be offered Tier 3 carrier screening for autosomal recessive conditions (Tables 1 – 5) when carrier screening is performed simultaneously with their partner.”
- “All XX patients should be offered screening for only those X-linked genes listed in Table 6 as part of Tier 3 screening.”
- “When Tier 1 or Tier 2 carrier screening was performed in a prior pregnancy, Tier 3 screening should be offered” (Gregg et al., 2021)
ACMG table 2 lists autosomal recessive genes for screening with carrier frequency and includes Fanconi anemia, complementation C in the list (Gregg et al., 2021):
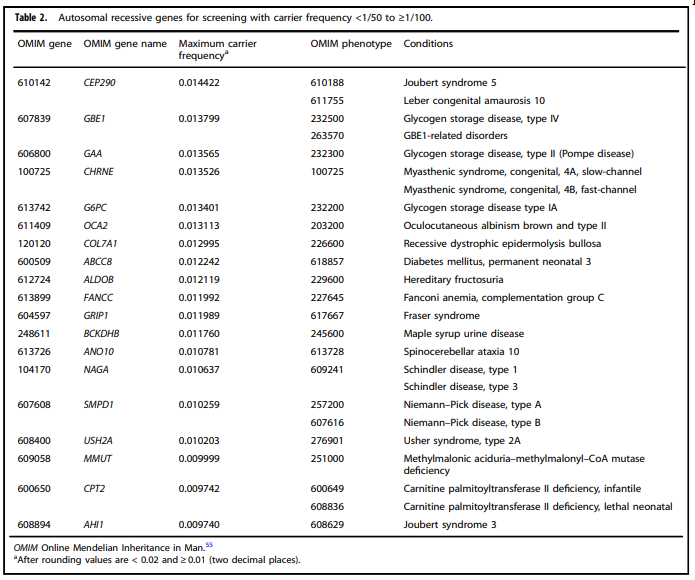
American College of Obstetricians and Gynecologists (ACOG) Committee Opinion on Carrier Screening for Genetic Conditions
In March 2017, ACOG issued a Committee Opinion on Carrier Screening for Genetic Conditions. ACOG recommends carrier screening and counseling before pregnancy; if results of screening are positive, ACOG recommends counseling the individual’s partner and family. ACOG further stipulates that screening for any particular condition should only be performed once. Finally, ACOG suggests that Ashkenazi individuals should consider screening for Fanconi anemia due to the higher-than-average carrier rates for that specific population (ACOG, 2017). ACOG guidelines were reaffirmed in 2020.
Second Pediatric Blood and Marrow Transplant Consortium International Conference on Late Effects after Pediatric HCT
Due to recent increase in survival following a hematopoietic cell transplant (HCT), the conference recommends continued screening and follow up with a wide variety of specialists, with focus on the late side-effects of HCT. The conference emphasizes the importance of screening for cancer due to the increased cumulative risk (Dietz et al., 2017).
The National Organization for Rare Disorders
NORD has published several recommendations for testing patients with suspected FA. These recommendations state that “FA should be suspected and tested for in any infant born with the thumb and arm abnormalities described previously. Anyone developing aplastic anemia at any age should be tested for FA, even if no other defects are present. Any patient who develops squamous cell carcinoma of the head and neck, gastrointestinal or gynecologic system at an early age with or without a history of tobacco or alcohol use, should be tested for FA. Many FA patients show no other abnormalities. It is essential to test for FA before contemplating stem cell transplantation for aplastic anemia or treatment for cancer, as standard chemotherapy and radiation protocols may prove toxic to FA patients” (NORD, 2020).
“Complementation testing is usually done first in order to identify which FA gene is mutated. Sequence analysis of the appropriate gene can then be done to determine the specific mutation in that gene. If a mutation is not identified, deletion/duplication analysis is available clinically for the genes associated with FA. Targeted mutation analysis is available for the common Ashkenazi Jewish FANCC mutation (NORD, 2020).”
Cancer Care Ontario
In December 2016, the CCO published recommendations for malignant hematology conditions. It is stated that patients with suspected aplastic anemia may be tested for FA via a peripheral blood chromosomal breakage analysis, such as the diepoxybutane test (DEB Test); further, all transplant candidates and siblings of FA patients with suspected aplastic anemia should be screened with this test (CCO, 2016).
The National Comprehensive Cancer Network
As FA often results in higher incidence of cancers, the NCCN has noted some observations regarding this condition. In the guideline for Esophageal and Esophagogastric Junction Cancers, the NCCN stated:
- The genes involved include FA complementation groups A-E, with FA-A (FANCA) located at 15q24.3; FA-B (FANCB), unknown; FA-C (FANCC) at 9q22.3; FA-D (FANCD) at 3p26-p22; and FA-E (FANCE), unknown.
- Mutations in FANCA and FANCC have been identified. Individuals are identified by pancytopenia and chromosome breakage and hematologic abnormalities, including anemia, bleeding, and easy bruising.
- Increased frequency of SCC of the esophagus as well as other squamous epithelium is observed.
- Karyotyping does not identify individuals with FA, but enhanced chromosome breakage with mitomycin C can identify homozygotes but not heterozygotes (NCCN, 2022).
United Kingdom National Multidisciplinary Guidelines
These recommendations were specifically made in the context of head and neck cancers. The recommendations for Fanconi anemia (FA) are as follows:
- “FA patients should receive prophylactic vaccination against high-risk HPV virus.
- FA patients should have quarterly screening for head and neck squamous cell carcinoma and an aggressive biopsy policy…treatment for head and neck squamous cell carcinoma with surgery alone where possible.”
- FA patients should follow up with a specialty Fanconi clinic (Shaw & Beasley, 2016).
U.S. Preventive Services Task Force (USPSTF) Recommendations
No U.S. Preventive Services Task Force recommendations for genetic testing for FA have been identified. A search for “Fanconi” on the USPSTF website turned up 0 results on October 19th, 2022.
Fanconi Anemia Research Fund
The Fanconi Anemia Research Fund released clinical care guidelines on diagnosis of Fanconi Anemia and provided a schematic representing a suggested algorithm for Fanconi anemia testing (Figure 1).
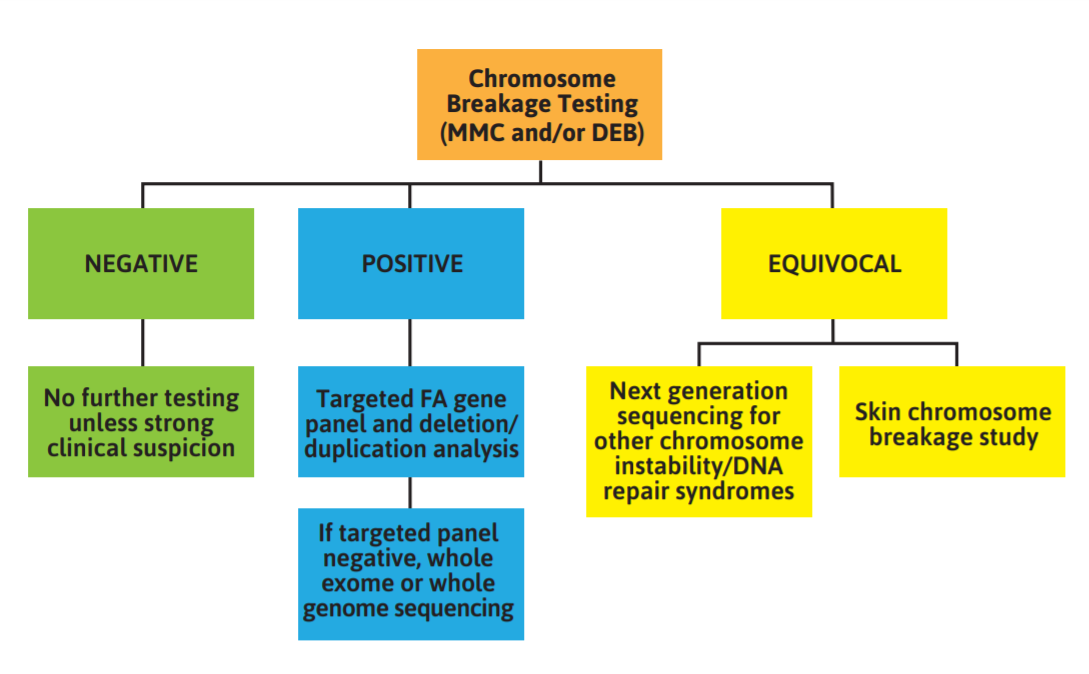
Figure 1: Schematic representing a suggested algorithm for Fanconi anemia testing
- “The gold-standard test for diagnosing Fanconi anemia (FA) is the chromosome breakage test (CBT) using the DNA cross-linking agents mitomycin C (MMC) and diepoxybutane (DEB). If a patient has a negative CBT, no further testing is necessary unless there is strong clinical suspicion. In this case, a skin cell CBT should be performed. If the CBT has a positive result, targeted FA gene panel should be performed. If the targeted panel is negative, whole exome or whole genome sequencing can be performed. An equivocal or inconclusive result will require next generation sequencing for variants that cause other chromosome instability syndromes, or a skin CBT for confirmation of FA.
- If the results from the chromosome breakage test are positive, genetic testing should be performed to identify the specific FA-causing variants.
- Following a positive chromosome breakage test, NGS panel testing for clinically available FA genes should be offered as the next step of testing. In addition to sequencing, testing should always include copy number analysis that will identify large deletions, duplications and insertions. Copy number variants (CNV) can be performed in tandem with panel testing or as a reflex test. In cases where the diagnosis of FA is in question, broader panels targeting a specific phenotype such as bone marrow failure or MDS/AML may be considered. Broad panels often are not comprehensive for each of the syndromes they analyze, so an FA-specific panel is still preferred when the diagnosis of FA is considered likely.
- Clinical WGS recently has been made available; however, the analysis largely remains focused on exons and splice sites, as the ability to interpret the impact of variants outside of those regions is still limited. The high cost of such testing currently prohibits this as a frontline testing tool. It may be warranted to use WES for an individual with a diagnosis of FA based on a positive chromosome breakage test but without causative variants identified on a dedicated FA panel test. While WES and WGS are beneficial for detecting variants in a larger area of the genome when compared to panel testing, these methods are not without risks and limitations.
- Following the diagnosis of FA, a cytogenetic study of the chromosomes of the patient’s bone marrow cells should be analyzed using standard G-banding methodology. If it becomes difficult to characterize using G-banding alone, fluorescence in situ hybridization (FISH), which employs fluorescently labeled chromosome region or gene-specific probes, can be a highly informative addition to G-banded chromosome analysis (FARF, 2020).”
References
- ACMG. (2021). STANDARDS AND GUIDELINES FOR CLINICAL GENETICS LABORATORIES. In A. C. o. M. G. a. Genomics (Ed.).
- ACOG. (2017). Carrier Screening for Genetic Conditions. https://www.acog.org/-/media/Committee-Opinions/Committee-on-Genetics/co691.pdf?dmc=1&ts=20170328T2033134535
- Alter, B. P. (2014). Fanconi anemia and the development of leukemia. Best Pract Res Clin Haematol, 27(3-4), 214-221. https://doi.org/10.1016/j.beha.2014.10.002
- Alter, B. P., & Giri, N. (2016). Thinking of VACTERL-H? Rule out Fanconi Anemia according to PHENOS. Am J Med Genet A, 170(6), 1520-1524. https://doi.org/10.1002/ajmg.a.37637
- Alter, B. P., Giri, N., McReynolds, L. J., & Altintas, B. (2022). Fanconi anaemia: A syndrome with distinct subgroups. Br J Haematol, 197(4), 467-474. https://doi.org/10.1111/bjh.18091
- Auerbach, A. D. (2015). Diagnosis of Fanconi anemia by diepoxybutane analysis. Curr Protoc Hum Genet, 85, 8.7.1-17. https://doi.org/10.1002/0471142905.hg0807s85
- Bogliolo, M., Pujol, R., Aza-Carmona, M., Munoz-Subirana, N., Rodriguez-Santiago, B., Casado, J. A., Rio, P., Bauser, C., Reina-Castillon, J., Lopez-Sanchez, M., Gonzalez-Quereda, L., Gallano, P., Catala, A., Ruiz-Llobet, A., Badell, I., Diaz-Heredia, C., Hladun, R., Senent, L., Argiles, B., . . . Surralles, J. (2019). Optimised molecular genetic diagnostics of Fanconi anaemia by whole exome sequencing and functional studies. J Med Genet. https://doi.org/10.1136/jmedgenet-2019-106249
- CCO. (2016). Consensus Pathology Recommendations for Complex Malignant Hematology. file:///C:/Users/AHCS6886/OneDrive%20-%20AVALON%20HEALTH%20SERVICES,%20LLC/Downloads/CMH_Appendix_C-ConsensusPathologyRecomm.pdf
- Chan, S. H., Ni, Y., Li, S.-T., Teo, J. X., Ishak, N. D. B., Lim, W. K., & Ngeow, J. (2021). Spectrum of Germline Mutations Within Fanconi Anemia–Associated Genes Across Populations of Varying Ancestry. JNCI Cancer Spectrum, 5(1). https://doi.org/10.1093/jncics/pkaa117
- Chang, L., Gao, X., Ji, G., Cheng, X., Zou, Y., Cheng, T., Yuan, W., & Zhu, X. (2021). Novel diagnostic approaches for Fanconi anemia (FA) by single-cell sequencing and capillary nano-immunoassay. Blood Science, 3(1), 20-25. https://doi.org/10.1097/bs9.0000000000000065
- Cheung, R. S., & Taniguchi, T. (2017). Recent insights into the molecular basis of Fanconi anemia: genes, modifiers, and drivers. Int J Hematol, 106(3), 335-344. https://doi.org/10.1007/s12185-017-2283-4
- D'Andrea, A. D. (2010). Susceptibility pathways in Fanconi's anemia and breast cancer. N Engl J Med, 362(20), 1909-1919. https://doi.org/10.1056/NEJMra0809889
- Dietz, A. C., Savage, S. A., Vlachos, A., Mehta, P. A., Bresters, D., Tolar, J., Bonfim, C., Dalle, J. H., de la Fuente, J., Skinner, R., Boulad, F., Duncan, C. N., Baker, K. S., Pulsipher, M. A., Lipton, J. M., Wagner, J. E., & Alter, B. P. (2017). Late Effects Screening Guidelines after Hematopoietic Cell Transplantation for Inherited Bone Marrow Failure Syndromes: Consensus Statement From the Second Pediatric Blood and Marrow Transplant Consortium International Conference on Late Effects After Pediatric HCT. Biol Blood Marrow Transplant, 23(9), 1422-1428. https://doi.org/10.1016/j.bbmt.2017.05.022
- FARF. (2020). Fanconi Anemia Clinical Care Guidelines, Fifth Edition. https://www.fanconi.org/images/uploads/other/Fanconi_Anemia_Clinical_Care_Guidelines_5thEdition_web.pdf
- Gregg, A. R., Aarabi, M., Klugman, S., Leach, N. T., Bashford, M. T., Goldwaser, T., Chen, E., Sparks, T. N., Reddi, H. V., Rajkovic, A., Dungan, J. S., Practice, A. P., & Guidelines, C. (2021). Screening for autosomal recessive and X-linked conditions during pregnancy and preconception: a practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med, 23(10), 1793-1806. https://doi.org/10.1038/s41436-021-01203-z
- Hays, L. (2014). Fanconi Anemia: Guidelines for Diagnosis and Management (4th ed.). Fanconi Anemia Research Fund. https://www.fanconi.org/images/uploads/other/FARF_Guidelines_Book_interior_lo-res.pdf
- Jung, M., Mehta, P. A., Jiang, C. S., Rosti, R. O., Usleaman, G., Correa da Rosa, J. M., Lach, F. P., Goodridge, E., Auerbach, A. D., Davies, S. M., Smogorzewska, A., & Boulad, F. (2020). Comparison of the clinical phenotype and haematological course of siblings with Fanconi anaemia. Br J Haematol. https://doi.org/10.1111/bjh.17061
- Kook, H., Cho, D., Cho, S. H., Hong, W. P., Kim, C. J., Park, J. Y., Yoon, W. S., Ryang, D. W., & Hwang, T. J. (1998). Fanconi anemia screening by diepoxybutane and mitomicin C tests in Korean children with bone marrow failure syndromes. J Korean Med Sci, 13(6), 623-628. https://doi.org/10.3346/jkms.1998.13.6.623
- Kottemann, M. C., & Smogorzewska, A. (2013). Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature, 493(7432), 356-363. https://doi.org/10.1038/nature11863
- Kutler, D. I., Patel, K. R., Auerbach, A. D., Kennedy, J., Lach, F. P., Sanborn, E., Cohen, M. A., Kuhel, W. I., & Smogorzewska, A. (2016). Natural history and management of Fanconi anemia patients with head and neck cancer: A 10-year follow-up. Laryngoscope, 126(4), 870-879. https://doi.org/10.1002/lary.25726
- Lei, L. C., Yu, V. Z., Ko, J. M. Y., Ning, L., & Lung, M. L. (2020). FANCD2 Confers a Malignant Phenotype in Esophageal Squamous Cell Carcinoma by Regulating Cell Cycle Progression. Cancers (Basel), 12(9). https://doi.org/10.3390/cancers12092545
- Longerich, S., Li, J., Xiong, Y., Sung, P., & Kupfer, G. M. (2014). Stress and DNA repair biology of the Fanconi anemia pathway. Blood, 124(18), 2812-2819. https://doi.org/10.1182/blood-2014-04-526293
- Milletti, G., Strocchio, L., Pagliara, D., Girardi, K., Carta, R., Mastronuzzi, A., Locatelli, F., & Nazio, F. (2020). Canonical and Noncanonical Roles of Fanconi Anemia Proteins: Implications in Cancer Predisposition. Cancers (Basel), 12(9). https://doi.org/10.3390/cancers12092684
- NCCN. (2022, Sept 2022). Esophageal and Esophagogastric Junction Cancers. Retrieved October 19, 2022 from https://www.nccn.org/professionals/physician_gls/pdf/esophageal.pdf
- NORD. (2020). Fanconi Anemia. https://rarediseases.org/rare-diseases/fanconi-anemia/
- Olson, T. S. (2022, 06/16/2022). Clinical manifestations and diagnosis of Fanconi anemia. https://www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-fanconi-anemia
- Rio, P., Navarro, S., Wang, W., Sanchez-Dominguez, R., Pujol, R. M., Segovia, J. C., Bogliolo, M., Merino, E., Wu, N., Salgado, R., Lamana, M. L., Yanez, R. M., Casado, J. A., Gimenez, Y., Roman-Rodriguez, F. J., Alvarez, L., Alberquilla, O., Raimbault, A., Guenechea, G., . . . Bueren, J. A. (2019). Successful engraftment of gene-corrected hematopoietic stem cells in non-conditioned patients with Fanconi anemia. Nat Med, 25(9), 1396-1401. https://doi.org/10.1038/s41591-019-0550-z
- Shaw, R., & Beasley, N. (2016). Aetiology and risk factors for head and neck cancer: United Kingdom National Multidisciplinary Guidelines. J Laryngol Otol, 130(S2), S9-s12. https://doi.org/10.1017/s0022215116000360
- Shimamura, A., & Alter, B. P. (2010). Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev, 24(3), 101-122. https://doi.org/10.1016/j.blre.2010.03.002
- Steinberg-Shemer, O., Goldberg, T. A., Yacobovich, J., Levin, C., Koren, A., Revel-Vilk, S., Ben-Ami, T., Kuperman, A. A., Shkalim Zemer, V., Toren, A., Kapelushnik, J., Ben-Barak, A., Miskin, H., Krasnov, T., Noy-Lotan, S., Dgany, O., & Tamary, H. (2019). Characterization and genotype-phenotype correlation of patients with Fanconi anemia in a multi-ethnic population. Haematologica. https://doi.org/10.3324/haematol.2019.222877
- Sumpter, R., Jr., & Levine, B. (2017). Emerging functions of the Fanconi anemia pathway at a glance. J Cell Sci, 130(16), 2657-2662. https://doi.org/10.1242/jcs.204909
- Sumpter, R., Jr., Sirasanagandla, S., Fernández Á, F., Wei, Y., Dong, X., Franco, L., Zou, Z., Marchal, C., Lee, M. Y., Clapp, D. W., Hanenberg, H., & Levine, B. (2016). Fanconi Anemia Proteins Function in Mitophagy and Immunity. Cell, 165(4), 867-881. https://doi.org/10.1016/j.cell.2016.04.006
- Yu, H., Pan, R., Gao, T., Wu, D., Ying, J., & Duan, S. (2020). FANCF hypomethylation is associated with colorectal cancer in Han Chinese. Turk J Gastroenterol, 31(8), 558-565. https://doi.org/10.5152/tjg.2020.19394
Coding Section
| Codes | Number | Description |
| CPT | ||
| 81242 | FANCC (fanconi anemia, complementation group C) (e.g., Fanconi anemia, type C) gene analysis, common variant (e.g., IVS4+4a>T) | |
| ICD-9 Procedure | ||
| ICD-9 Diagnosis | 284.09 | Other constitutional aplastic anemia (includes Fanconi anemia) |
| 284.9 | Aplastic anemia, unspecified (includes bone marrow failure) | |
| HCPCS | ||
| ICD-10-CM (effective 10/01/15) | D61.09 | Other constitutional aplastic anemia (includes Fanconi anemia) |
| D61.89 | Other specified aplastic anemias and other bone marrow failure syndromes | |
| D61.9 | Aplastic anemia, unspecified | |
| F70 – F79 | Intellectual Disabilities | |
| H02.0 – H02.1 | Other disorders of eyelid | |
| H57.8 | Other specified disorders of eye | |
| L81.3 & L81.8 | Café au lait spots and Other disorders of pigmentation | |
| M40 – M43 | Deforming dorsopathies | |
| N28.89 | Other specified disorders of kidney | |
| P07.00 – P07.18 | Low birth weight newborn | |
| Q17.3 | Other misshapen ear | |
| R53.8 | Fatigue | |
| ICD-10-PCS (effective 10/01/15) | Not applicable, ICD-10-PCS codes are only used for inpatient services. There are no ICD procedure codes for labortory. | |
| Type of Service | ||
| Place of Service |
Procedure and diagnosis codes on Medical Policy documents are included only as a general reference tool for each policy. They may not be all-inclusive.
This medical policy was developed through consideration of peer-reviewed medical literature generally recognized by the relevant medical community, U.S. FDA approval status, nationally accepted standards of medical practice and accepted standards of medical practice in this community, Blue Cross Blue Shield Association technology assessment program (TEC) and other non-affiliated technology evaluation centers, reference to federal regulations, other plan medical policies, and accredited national guidelines.
"Current Procedural Terminology© American Medical Association. All Rights Reserved"
History From 2014 Forward
| 01/25/2023 | Annual review, updating policy for clarity and consistency. removing subcriteria for prenatal/carrier testigg Also updating description, rationale and references. |
|
01/14/2022 |
Annual review, updating policy criteria #2, also updating rationale and references. |
|
01/04/2021 |
Annual review, no change to policy intent. Updating description, rationale and references. |
|
01/02/2020 |
Annual review, no change to policy intent. |
|
06/19/2019 |
Interim review. Genetic counseling is recommended is replacing Genetic counseling is Medically necessary. No other changes made. |
|
01/02/2019 |
Annual review, no change to policy intent. |
|
02/20/2018 |
Annual review, updating policy to include genetic counseling, also updating background, description, rationale and references. |
|
04/26/2017 |
Interim review to align with Avalon quarterly schedule. Updated category to Laboratory. |
|
03/07/2017 |
Updated codes in coding section. |
|
12/05/2016 |
Annual review, no change to policy intent. |
|
12/16/2015 |
Annual review, no change to policy intent. Updating background, description, rationale and references. Adding appendix 1. |
|
12/23/2014 |
New Policy. |