
Genetic Testing for the Diagnosis of Inherited Peripheral Neuropathies - CAM 299
Description
The inherited peripheral neuropathies are a heterogeneous group of diseases that may be inherited in an autosomal dominant, autosomal recessive, or X-linked dominant manner. The inherited peripheral neuropathies can be divided into hereditary motor and sensory neuropathies (such as Charcot-Marie-Tooth disease), hereditary neuropathy with liability to pressure palsies, hereditary sensory and autonomic neuropathies, and other miscellaneous types (e.g., hereditary brachial plexopathy, giant axonal neuropathy). In addition to clinical presentation, nerve conduction studies, and family history, genetic testing can be used to diagnose specific inherited peripheral neuropathies (Kang, 2022a).
Policy
- Genetic Counseling is considered MEDICALLY NECESSARY and is recommended for genetic testing of Charcot-Marie-Tooth (CMT) disease or other inherited peripheral neuropathies.
- Genetic Testing for CMT disease is considered MEDICALLY NECESSARY when the patient displays clinical features of CMT and a definitive diagnosis remains uncertain after history, physical examination, genetic counseling, and completion of conventional diagnostic studies (i.e., nerve conduction studies and/or electromyography). If results indicate a demyelinating neuropathy, the most commonly identified CMT subtype, CMT1A (PMP22 duplication), should be tested for first.
- Genetic testing for CMT in asymptomatic at-risk individuals is considered MEDICALLY NECESSARY if there is a close relative (i.e., first, second, or third degree relative) with a known CMT mutation.
- Genetic testing of known familial CMT mutation(s) is considered MEDICALLY NECESSARY for prenatal diagnosis in at-risk pregnancies.
- When clinical features are significantly suggestive of CMT and the genetic tests are negative, peripheral nerve biopsy is considered MEDICALLY NECESSARY to diagnose CMT.
- Genetic testing for hereditary neuropathy with liability to pressure palsies (HNPP) (PMP22 deletion) is considered MEDICALLY NECESSARY when the patient displays clinical features of HNPP and a definitive diagnosis remains uncertain after history, physical examination, genetic counseling, and completion of electrophysiologic studies.
- Genetic testing for hereditary motor neuropathy (HMN) (BSCL2 gene) is considered MEDICALLY NECESSARY when the patient displays clinical features of HMN and a definitive diagnosis remains uncertain after history, physical examination, genetic counseling, and completion of electrophysiologic studies.
NOTE 1: For all other uncommon hereditary peripheral neuropathy gene testing, refer to CAM 166 General Genetic Testing, Germline Disorders.
NOTE 2: For 5 or more gene tests being run on the same platform, such as multi-gene panel next generation sequencing, please refer to CAM 235.
Table of Terminology
|
Term |
Definition |
|
AAN |
American Academy of Neurology |
|
AANEM |
American Academy of Neuromuscular and Electrodiagnostic Medicine |
|
AAPM&R |
American Academy of Physical Medicine and Rehabilitation |
|
AARS |
Alanine-tRNA ligase, cytoplasmic |
|
AIFM1 |
Apoptosis-inducing factor 1 |
|
BSCL2 |
Seipin lipid droplet biogenesis associated |
|
DYNC1H1 |
Dynein cytoplasmic 1 heavy chain 1 |
|
EFNS |
European Federation of Neurological Societies |
|
EGR2 |
Early growth response protein 2 |
|
GARS |
Glycyl-t-ribonucleic acid synthetase |
|
GDAP1 |
Ganglioside-induced differentiation-associated protein-1 |
|
GJB1 |
Gap junction beta-1 protein (connexin 32) |
|
HMN |
Hereditary motor neuropathy |
|
HNPP |
Hereditary neuropathy with predisposition to pressure palsy |
|
HSANs |
Hereditary sensory and autonomic neuropathies |
|
HSPB1 |
Heat-shock protein beta-1 |
|
HSPB8 |
Heat-shock protein beta-8 |
|
KIF1B |
Kinesin family member 1B |
|
LITAF |
Lipopolysaccharide induced tumor necrosis factor |
|
LMNA |
Lamin A/C |
|
MED25 |
Mediator complex subunit 25 |
|
MPZ |
Myelin protein P0 |
|
NEFL |
Neurofilament light polypeptide |
|
NINDS |
National Institute of Neurological Disorders and Stroke |
|
PDK3 |
Pyruvate dehydrogenase kinase isoform 3 |
|
PMP22 |
Peripheral myelin protein 22 |
|
PRPS1 |
Ribose-phosphate pyrophosphokinase 1 |
|
RAB7A |
Member RAS oncogene family |
|
Sep T9 |
Septin 9 |
|
SH3TC2 |
SH3 domain and tetratricopeptide |
|
TRPV4 |
Transient receptor potential cation channel subfamily V member 4 |
|
WES/WGS |
Whole exome or genome sequencing |
Rationale
Peripheral neuropathies encompass the set of disorders that primarily lead to peripheral nerve dysfunction. Symptoms typically include weakness of muscles at extremities, spine curvature, and loss of sensation at extremities (Kang, 2019; UTD, 2021). Neuropathies may be caused by a variety of different factors, such as metabolic issues (including Fabry disease, Niemann-Pick disease, et al.) or present as a secondary symptom to another condition (such as Tangier disease) (Kang, 2019).
Charcot-Marie-Tooth (CMT) disease, also known as hereditary motor sensory neuropathy, is a group of progressive disorders that affect the peripheral nerves. CMT is caused by a mutation in one of several myelin genes that result in defects in myelin structure, maintenance, or function within peripheral nerves. Charcot-Marie-Tooth disease is one of the most common inherited neurological disorders, affecting approximately 1 in 2,500 people in the United States (Kang, 2022a).
Symptoms
The neuropathy of CMT affects both motor and sensory nerves. Symptoms usually start in childhood and have a gradual progression. The severity of symptoms varies greatly among individuals and even among family members with the disease (Bird, 2020; NINDS, 2007). Typical symptoms include the following:
- Weakness of the foot and lower leg muscles, which may result in foot drop and a high-stepped gait with frequent ankle sprains, tripping or falls
- Foot deformities, such as pes cavus and hammertoes
- Distal calf muscle atrophy often occurs, causing the stork leg deformity or inverted champagne bottle appearance
- Weakness and muscle atrophy may occur in the hands, resulting in difficulty with carrying out fine motor skills.
- Sensory loss is gradual and mainly involves proprioception and vibration.
- Spinal deformities like kyphosis and scoliosis can often develop (NINDS, 2007)
Pain can range from mild to severe, and some people may need to rely on foot or leg braces or other orthopedic devices to maintain mobility. Although in rare cases, individuals may have respiratory muscle weakness, CMT is not considered a fatal disease and people with most forms of CMT have a normal life expectancy (NINDS, 2007).
Causes
CMT is caused by mutations in genes that produce proteins involved in the structure and function of either the peripheral nerve axon or the myelin sheath. Although different proteins are abnormal in different forms of CMT disease, all mutations affect the normal function of the peripheral nerves. There is little correlation between the genotype and phenotype of CMT; it is common to see differing mutations result in various clinical phenotypes all within the same gene (Kang, 2022a).
Pattern of Inheritance
The pattern of inheritance varies with the type of CMT disease. CMT1, most cases of CMT2, and most intermediate forms are inherited in an autosomal dominant pattern. CMT4, a few CMT2 subtypes, and some intermediate forms are inherited in an autosomal recessive pattern. CMTX is inherited in an X-linked pattern. In the X-linked recessive patterns, only males develop the disease, although females who inherit the defective gene can pass the disease onto their sons. In the X-linked dominant pattern, an affected mother can pass on the disorder to both sons and daughters, while an affected father can only pass it onto his daughters. Some cases of CMT disease result from a new mutation and occur in people with no history of the disorder in their family. In rare cases the gene mutation causing CMT disease is a new mutation which occurs spontaneously in the individual's genetic material and has not been passed down through the family (Kang, 2022a).
CMT1
|
Locus Name |
Proportion of CMT1 (excluding CMTX) |
Gene |
Protein Product |
|
CMT1A |
70% – 80% |
PMP22 |
Peripheral myelin protein 22 |
|
CMT1B |
10% – 12% |
MPZ |
Myelin protein P0 |
|
CMT1C |
~ 1% |
LITAF |
Lipopolysaccharide-induced tumor necrosis factor-alpha factor |
|
CMT1D |
Unknown |
EGR2 |
Early growth response protein 2 |
|
CMT1E |
~ 1% |
PMP22 |
Peripheral myelin protein 22 |
|
CMT1F/2E |
Unknown |
NEFL |
Neurofilament light polypeptide |
CMT1A is an autosomal dominant disease that results from a duplication of the gene on chromosome 17 that carries the instructions for producing the peripheral myelin protein-22 (PMP-22). Overexpression of this gene causes the structure and function of the myelin sheath to be abnormal. A different neuropathy distinct from CMT1A called hereditary neuropathy with predisposition to pressure palsy (HNPP) is caused by a deletion of one of the PMP-22 genes. In this case, abnormally low levels of the PMP-22 gene result in episodic, recurrent demyelinating neuropathy (NINDS, 2007).
CMT1B is an autosomal dominant disease caused by mutations in the gene that carries the instructions for manufacturing the myelin protein zero (P0), which is another critical component of the myelin sheath. Most of these mutations are point mutations. As a result of abnormalities in P0, CMT1B produces symptoms similar to those found in CMT1A (NINDS, 2007).
The less common CMT1C, CMT1D, and CMT1E, which also have symptoms similar to those found in CMT1A, are caused by mutations in the LITAF, EGR2, and NEFL genes, respectively (NINDS, 2007).
CMT2
CMT2 is an axonal (non-demyelinating) peripheral neuropathy characterized by distal muscle weakness and atrophy. Nerve conduction velocities are usually within the normal range; however, occasionally they fall in the low-normal or mildly abnormal range (Bird, 2019). In general, individuals with CMT2 tend to be less disabled and have less sensory loss than individuals with CMT1 (Bird, 2019). It is less common than CMT1. CMT2A, the most common axonal form of CMT, is caused by mutations in Mitofusin 2, a protein associated with mitochondrial fusion. CMT2A has also been linked to mutations in the gene that codes for the kinesin family member 1B-beta protein, but this has not been replicated in other cases. Other less common forms of CMT2 are associated with various genes: CMT2B (associated with RAB7), CMT2D (GARS). CMT2E (NEFL), CMT2H (HSP27), and CMT2l (HSP22) (NINDS, 2007).
Table 2: Molecular Genetics of CMT2 (Bird, 2019)
|
Locus |
Proportion of CMT |
Gene / Chromosome Locus |
Protein Product |
|
|
CMT2A1 |
Unknown |
KIF1B |
Kinesin-like protein KIF1B |
|
|
CMT2A21 |
20% |
MFN2 |
Mitofusin-2 |
|
|
CMT2B |
Unknown |
RAB7A |
Ras-related protein Rab-7 |
|
|
CMT2B1 |
Unknown |
LMNA |
Lamin A/C |
|
|
CMT2B2 |
Unknown |
MED25 |
Mediator of RNA polymerase II transcription subunit 25 |
|
|
CMT2C2 |
Unknown |
TRPV4 |
Transient receptor potential cation channel subfamily V member 4 |
|
|
CMT2D3 |
3% |
GARS |
Glycyl-tRNA synthetase |
|
|
CMT2E/1F4 |
4% |
NEFL |
Neurofilament light polypeptide |
|
|
CMT2F |
Unknown |
HSPB1 |
Heat-shock protein beta-1 |
|
|
CMT2G |
Unknown |
12q12-q13 |
Unknown |
|
|
CMT2H/2K |
5% |
GDAP1 |
Ganglioside-induced differentiation-associated protein-1 |
|
|
CMT2I/2J |
Unknown |
MPZ |
Myelin protein P0 |
|
|
CMT2L |
Unknown |
HSPB8 |
Heat-shock protein beta-8 |
|
|
CMT2N |
Unknown |
AARS |
Alanine--tRNA ligase, cytoplasmic |
|
|
CMT2O |
Unknown |
DYNC1H1 |
Cytoplasmic dynein 1 heavy chain 1 |
|
|
CMT2P |
Unknown |
LRSAM1 |
E3 ubiquitin-protein ligase LRSAM1 |
|
|
CMT2S |
Unknown |
IGHMBP2 |
DNA-binding protein SMUBP-2 |
|
|
CMT2T |
Unknown |
DNAJB2 |
DnaJ homolog subfamily B member 2 |
|
|
CMT2U |
Unknown |
MARS |
Methionine--tRNA ligase, cytoplasmic |
|
|
1. (Züchner, 2013). 2. (Schindler, 2014). 3. (Anthony Antonellis, 2018). 4. (Peter De Jonghe, 2011) |
||||
CMT3
CMT3, or Dejerine-Sottas disease, is a severe demyelinating neuropathy that begins in infancy. Infants have severe muscle atrophy, weakness, and sensory problems. This rare disorder can be caused by a specific point mutation in the P0 gene or a point mutation in the PMP-22 gene (NINDS, 2007).
CMT4
CMT4 comprises several different subtypes of autosomal recessive demyelinating motor and sensory axonal neuropathies. Each neuropathy subtype is caused by a different genetic mutation, may affect a particular ethnic population, and produces distinct physiologic or clinical characteristics. Affected individuals have the typical CMT phenotype of distal muscle weakness and atrophy associated with sensory loss and, frequently, pes cavus foot deformity. Several genes have been identified as causing CMT4, including GDAP1 (CMT4A), MTMR13 (CMT4B1), MTMR2 (CMT4B2), SH3TC2 (CMT4C), NDG1 (CMT4D), EGR2 (CMT4E), PRX (CMT4F), FDG4 (CMT4H), and FIG4 (CMT4J) (Kang, 2022a; NINDS, 2007).
Table 3: Molecular Genetics of CMT4 (Bird, 2019)
|
Locus Name |
Proportion of CMT4 |
Gene |
Protein Product |
|
CMT4A1 |
Unknown |
GDAP1 |
Ganglioside-induced differentiation-associated protein 1 |
|
CMT4B1 |
MTMR2 |
Myotubularin-related protein 2 |
|
|
CMT4B2 |
SBF2 |
Myotubularin-related protein 13 |
|
|
CMT4C2 |
SH3TC2 |
SH3 domain and tetratricopeptide repeats-containing protein 2 |
|
|
CMT4D |
NDRG1 |
Protein NDRG1 |
|
|
CMT4E |
EGR2 |
Early growth response protein 2 |
|
|
CMT4F |
PRX |
Periaxin |
|
|
CMT4H3 |
FGD4 |
FYVE, RhoGEF and PH domain-containing protein 4 |
|
|
CMT4J4 |
FIG4 |
Phosphatidylinositol 3, 5 biphosphate |
|
|
1. (Bird, 2017) 2. (Hamid Azzedine, 2015) 3. (Delague, 2013) 4. (Li, 2013) |
|||
CMTX
CMTX is caused by a point mutation in the connexin-32 gene on the X chromosome. The connexin-32 protein is expressed in Schwann cells, which wrap around nerve axons and make up a single segment of the myelin sheath (NINDS, 2007). CMTX type 1 is characterized by a moderate to severe motor and sensory neuropathy although symptoms tend to be less severe in women. Hearing loss and central nervous system symptoms may also occur in certain affected families (Abrams, 2020).
Table 4: Molecular Genetics of CMTX
|
Disease Name |
Proportion of X-Linked CMT |
Gene / Chromosome Locus |
Protein Product |
|
CMTX11 |
90% |
GJB1 |
Gap junction beta-1 protein (connexin 32) |
|
CMTX22 |
Unknown |
Xp22.2 |
|
|
CMTX31 |
|
Not applicable |
|
|
CMTX41 |
AIFM1 |
Apoptosis-inducing factor 1 |
|
|
CMTX52 |
PRPS1 |
Ribose-phosphate pyrophosphokinase 1 |
|
|
CMTX61 |
PDK3 |
Pyruvate dehydrogenase kinase isoform 3 |
|
|
1. (Bird, 2020) 2. (Kim, 2013). |
|||
Hereditary Brachial Plexopathy (Hereditary Neuralgic Amyotrophy)
This condition is primarily characterized by painful injuries to the brachial plexus nerves as well as episodic weakness of the shoulder and arm. Other symptoms such as winging of the scapula, short stature, neck folds, small face, and hypotelorism may be present. Nerve conduction velocity is typically normal, and the histopathology of this condition is non-specific. The septin 9 gene (SEPT9) on chromosome 17 has been associated with this condition (Bromberg, 2021).
Giant Axonal Neuropathy
This condition is characterized by disorganization of cytoskeletal intermediate filaments stemming from a mutated form of gigaxonin. Patients with this disorder often have a signature physical appearance; red and kinked hair, high foreheads, long eyelashes, and pale complexions are all hallmarks of this condition. The central nervous system may be affected as well with cerebellar dysfunction, spasticity, and potentially intellectual disability as possible symptoms. Nerve biopsy may show axonal loss or other axonal dysfunction. This diagnosis is confirmed by testing of the GAN gene (Kang, 2019).
Hereditary Sensory and Autonomic Neuropathies (HSANs)
This subsection of disorders primarily encompasses non-motor neuropathies and are characterized by major loss of myelinated and unmyelinated fibers. These conditions are not as common as hereditary motor neuropathies and primarily present with sensory dysfunction, although motor functions may be affected. There are five main types of HSAN, each caused by different genes. Genes are associated as shown below (Eichler, 2021):
|
Disease Name (subtype) |
Gene(s) or Locus |
Examples of symptoms |
|
HSAN1 (A) |
SPTLC1 |
Distal sensory loss, distal muscle wasting |
|
HSAN1 (B) |
3p24-p22 |
Axonal neuropathy with distal sensory impairment |
|
HSAN1 (C) |
SPTLC2 |
Distal sensory loss, distal muscle wasting |
|
HSAN1 (D) |
ATL1 |
Distal sensory loss, distal muscle wasting |
|
HSAN1 (E) |
DNMT1 |
Hearing loss, progressive dementia |
|
HSAN1 (F) |
ATL3 |
Distal sensory impairment |
|
HSAN2 (A) |
HSN2 |
Loss of pain, pressure, touch, and temperature sensation |
|
HSAN2 (B) |
FAM134B |
Loss of pain, pressure, touch, and temperature sensation |
|
HSAN2 (C) |
KIF1A |
Loss of pain, pressure, touch, and temperature sensation |
|
HSAN2 (D) |
SCN9A |
Loss of pain and temperature sensation, hearing loss |
|
HSAN3/Familial Dysautonomia |
9q31 |
Dysautonomic crises, orthostatic hypotension |
|
HSAN4/Congenital Insensitivity to Pain with Anhidrosis |
NTRK1 |
Loss of pain sensation, thermoregulatory dysfunction |
|
HSAN5 |
NGFB |
Loss of pain and temperature sensation |
|
HSAN6 |
DST |
Lack of psychomotor development, respiratory difficulties |
|
HSAN7 |
SCN11A |
Inability to experience pain |
Other unclassified HSANs exist, such as spastic paraplegia with ulcerations of the hands and feet (associated with CCT5) and sensory neuropathy with ichthyosis and anterior chamber syndrome (Eichler, 2021).
Genetic Testing
Charcot-Marie-Tooth disease is usually diagnosed by an extensive history and physical examination. The clinical diagnosis is then confirmed by electrodiagnostic tests like electromyography and nerve conduction velocity tests, and sometimes by nerve biopsy. Genetic testing is available for most types of CMT, and results are usually enough to confirm a diagnosis. Genetic testing can simplify the diagnosis of CMT by avoiding invasive procedures, such as nerve biopsy. In addition, early diagnosis can facilitate early interventions, including physical therapy. However, most therapies are only supportive (occupational, physical) and generally do not rely on the results of specific genetic testing (Kang, 2022a, 2022b). A positive genetic test can confirm diagnosis in most people with CMT. But a negative result does not exclude the disease, as an unidentified gene may be missed by DNA sampling (News, 2021).
Genetic testing for CMT is complicated by the extensive underlying genetic heterogeneity. The CMT spectrum of disorders can be inherited in an autosomal dominant, autosomal recessive, or X-linked manner. The most commonly identified CMT subtypes are CMT1A (PMP22 duplication), CMTX1 (GJB1 mutation), hereditary neuropathy with liability to pressure palsies (PMP22 deletion), CMT1B (MPZ mutation), and CMT2A (MFN2 mutation). Together, these five subtypes account for 92 percent of genetically defined CMT cases. All other CMT subtypes and associated mutations each account for <1 percent of genetically defined CMT (CMTA; Kang, 2022a). Genetic screening for relatives of a patient diagnosed with CMT is an option, but risk assessment depends on several factors, including accuracy of the diagnosis, determination of the mode of inheritance for the individual family, and results of molecular genetic testing (Kang, 2022a). Numerous genetic panels are available for the assessment of peripheral neuropathies, such as GeneDx’s panel (64 genes) and Invitae’s panel (83 genes) (GeneDx, 2018; Invitae, 2020). Other pane’s include ones by Athena Diagnostics (23 genes) (Athena_Diagnostics, 2021), Claritas Genomics (PMP22 gene) (Claritas_Genomics, 2021), MNG Laboratories (139 genes) (MNG_Laboratories, 2021), Prevention Genetics (44 genes) (Prevention_Genetics, 2021), and Variantyx’s Genomic Unity panel (25 genes).
Clinical Utility and Validity
DiVincenzo et al. (2014) performed an analysis of the genetic landscape of CMT. 14 genes associated with CMT (PMP22, GJB1, MPZ, MFN2, SH3TC2, GDAP1, NEFL, LITAF, GARS, HSPB1, FIG4, EGR2, PRX, and RAB7A) were evaluated out of 3312 individuals. Deletions and duplications in the PMP22 gene consisted of about 78% of positive findings, followed by mutations in the GJB1 (6.7%), MPZ (5.3%), and MFN2 (4.3%) genes. 71% of the pathogenic mutations found were missense mutations. Overall, 95% of the positive results involved one of four genes (PMP22, GJB1, MPZ, MFN2). The authors conclude that these four genes should be screened first before proceeding with further genetic testing (DiVincenzo et al., 2014).
Pareyson (2017) reviewed the current literature on CMT diagnosis stating that data justifies a step-wise algorithm considering a variety of factors, such as phenotype, nerve conduction velocities, and ethnicity. The authors note that NGS is steadily replacing older methods of sequencing in this algorithm. The authors propose evaluating the first few common genes (PMP22, MPZ, et al) and then considering larger sequencing methods such as NGS. However, due to the growing number of genes associated with CMT, these larger sequencing methods may be considered first-line. Finally, the authors state that due to the growing number of associated genes, newer classifications need to be discussed and validated further (Pareyson et al., 2017).
Rudnik-Schöneborn and colleagues (2016) evaluated the clinical features and genetic results of 1206 CMT patients and 124 affected relatives. Genetic detection rates were 56% in demyelinating CMT and 17% in axonal CMT. “Three genetic defects (PMP22 duplication/deletion, GJB1/Cx32 or MPZ/P0 mutation) were responsible for 89.3% of demyelinating CMT index patients in whom a genetic diagnosis was achieved, and the diagnostic yield of the three main genetic defects in axonal CMT (GJB1/Cx32, MFN2, MPZ/P0 mutations) was 84.2%”. The authors concluded that “diagnostic algorithms are still useful for cost-efficient mutation detection and for the interpretation of large-scale genetic data made available by next generation sequencing strategies” (Rudnik-Schoneborn et al., 2016).
Vaeth et al. (2019) evaluated the effect of implementing a targeted next-generation sequencing (NGS) approach for identifying CMT. The authors stated that from 1992-2012, a total of 1442 CMT analyses were performed (through Sanger sequencing and other quantitative analyses) and a pathogenic variant was discovered in 21.6% of these cases. From this cohort, 195 samples that did not reach a definitive diagnosis were sequenced by a custom 63-gene panel. The authors identified a 5.6% increase in diagnostic yield using this targeted NGS approach (Vaeth et al., 2019).
Cortese et al. (2020) investigated the effectiveness of NGS panels in CMT. 220 patients were enrolled in the study and a targeted CMT NGS panel was performed. After NGS sequencing, a molecular diagnosis based on a pathogenic variant was found in 30% of the cases and variants of unknown significance were found in 33% of the cases. 39% of the cases held mutations in GJB1, MFN2, and MPZ while the others held mutations in SH3TC2, GDAP1, IGHMBP2, LRSAM1, FDG4, and GARS. Copy number changes were detected in PMP22, MPZ, MFN2, SH3TC2, and FDG4. The authors conclude that "NGS panels are effective tools in the diagnosis of CMT, leading to genetic confirmation in one-third of cases negative for PMP22 duplication/deletion, thus highlighting how rarer and previously undiagnosed subtypes represent a relevant part of the genetic landscape of CMT” (Cortese et al., 2020).
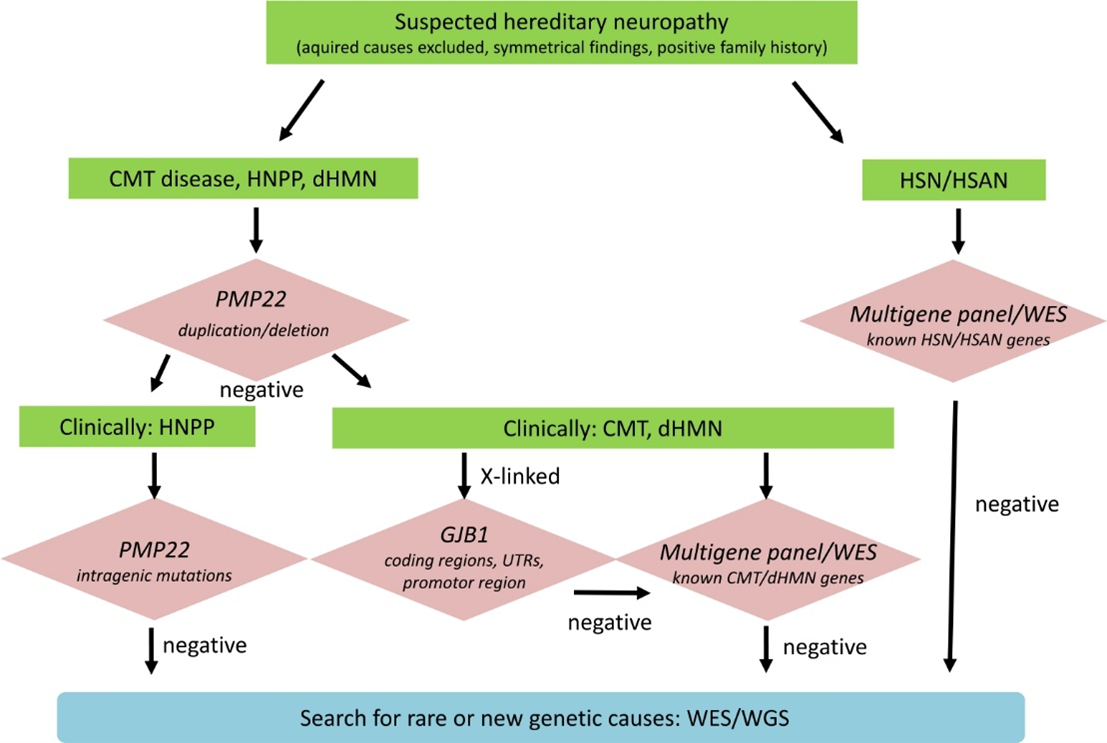
Rudnik-Schöneborn, Auer-Grumbach, and Senderek (2020) suggested a diagnostic algorithm for genetic testing of suspected hereditary neuropathy. Advanced genetic sequencing allows for comprehensive evaluation of the pathogenic relevance of identified variants. As shown in the chart above, “If PMP22 copy number analysis is negative, then clinical distinction of HNPP and CMT/dHMN will sort out patients for PMP22 mutation analysis only and those for broader multigene testing. If a pedigree is compatible with X-linked inheritance, it is recommended to analyze coding and non-coding regions of GBJ1. Patients who are tested negative for known neuropathy genes may be included in further whole exome or genome sequencing (WES/WGS) to detect mutations in rare and new genes” (Rudnik-Schöneborn et al., 2020).
Yalcintepe et al. (2021) studied the importance of multiple gene analysis for diagnosis of Charcot Marie Tooth Disease. Fifty-five patients with suspected CMT phenotype were examined using a customized multigene panel which was compared to the Multiplex Ligand Probe Amplification (MLPA) method. The custom panel identified 13 cases (7.15%) with a pathogenic/likely pathogenic variant. “The affected genes were MARS1, NDRG1, GJB1, GDAP1, MFN2, PRX, SH3TC2, and FGD4. Six cases (10.9%) had pathogenic variants in GJB1 and FGD4 genes, variants of unknown significance (VUS) in HSPB3, CHRNA1, ARHGEF10, and KIF5A genes. 21 cases (11.55%) had VUS with the genes HSPB3, KIF1B, SCN11A, CHRNA1, HSPB1, FIG4, ARHGEF10, DHTKD1, SBF1, EGR2, SBF2, IGHMBP2, KIF5A, and DNAJB2.” The authors concluded that the NGS customized panel was beneficial, time-saving, and cost-effective in the diagnosis of CMT (Yalcintepe et al., 2021).
AAN, AANEM, and AAPM&R
The Polyneuropathy Task Force that included 19 physicians with representatives from the American Academy of Neurology (AAN), the American Academy of Neuromuscular and Electrodiagnostic Medicine (AANEM), and the American Academy of Physical Medicine and Rehabilitation (AAPM&R) concluded that “genetic testing is established as useful for the accurate diagnosis and classification of hereditary polyneuropathies (Class I)” (England et al., 2009).
The Task Force stated that “for patients with a cryptogenic polyneuropathy who exhibit a classic hereditary neuropathy phenotype, routine genetic screening may be useful for CMT1A duplication/deletion and Cx32 mutations in the appropriate phenotype (Class III). Further genetic testing may be considered guided by the clinical question.” The Task Force recommended that “genetic testing should be conducted for the accurate diagnosis and classification of hereditary neuropathies (Level A)”. The Task force further recommended that “Genetic testing may be considered in patients with a cryptogenic polyneuropathy and classic hereditary neuropathy phenotype (Level C). There is insufficient evidence to support or refute the usefulness of routine genetic testing in cryptogenic polyneuropathy patients without a classic hereditary phenotype (Level U)” (England et al., 2009).
These guidelines were reaffirmed on Jan. 22, 2022.
European Federation of Neurological Societies (EFNS)
The EFNS released recommendations on genetic testing for various types of peripheral neuropathies. Regarding CMT, they noted that “Given the rarity of AR CMT in the European population routine diagnostic screening of the many known genes is currently not feasible” but acknowledged that “Currently, molecular genetic testing can be offered for several of the more prevalent CMT genes”. EFNS stated that PMP22 duplication should be tested first in patients presenting with CMT1, followed by sequencing of GJB1 (in case no male-to-male transmission is present), MPZ, and PMP22. If a patient presents with CMT2, MFN2 should be screened first, followed by MPZ. If a patient presents with intermediate CMT, GJB1 and MPZ should be screened. EFNS notes that in patients with hereditary neuropathy with liability to pressure palsies will be investigated for a PMP22 deletion at the same time as a screening for a PMP22 duplication (Burgunder et al., 2011).
However, routine diagnostic screenings for hereditary motor neuropathy (HMN) and hereditary sensory-autonomic neuropathy (HSAN) are not feasible due to low mutation frequencies. If screening is performed for these conditions, EFNS recommends BSCL2 as the first candidate for screening in HMN. NTRK1 may also be screened for in congenital insensitivity to pain with anhidrosis patients (CIPA, a sub-phenotype of HSAN) and RAB7 may be screened in CMT2B patients. Finally, SEPT9 may be screened in the context of hereditary neuralgic amyotrophy (HNA) (Burgunder et al., 2011).
Regulatory Status
A search on the FDA website for “neuropathy” on April 22, 2021 yielded no results pertaining to genetic testing (FDA, 2021). Additionally, many labs have developed specific tests that they must validate and perform in house. These laboratory-developed tests (LDTs) are regulated by the Centers for Medicare & Medicaid Services (CMS) as high-complexity tests under the Clinical Laboratory Improvement Amendments of 1988 (CLIA ’88). As an LDT, the U.S. Food and Drug Administration has not approved or cleared this test; however, FDA clearance or approval is not currently required for clinical use.
References
- Abrams, C. (2020). GJB1 Disorders: Charcot Marie Tooth Neuropathy (CMT1X) and Central Nervous System Phenotypes. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK1374/
- Anthony Antonellis, P., Lev G Goldfarb, MD, and Kumaraswamy Sivakumar, MD. (2018). GARS-Associated Axonal Neuropathy. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK1242/
- Athena_Diagnostics. (2021). CMT Advanced Evaluation - Comprehensive.
- Bird, T. (2017). GDAP1-Related Hereditary Motor and Sensory Neuropathy. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK1539/
- Bird, T. (2019). Charcot-Marie-Tooth (CMT) Hereditary Neuropathy Overview. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK1358/
- Bird, T. (2020). Charcot-Marie-Tooth (CMT) Hereditary Neuropathy Overview. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK1358/
- Bromberg, M. (2021). Brachial plexus syndromes. Retrieved from https://www.uptodate.com/contents/brachial-plexus-syndromes?sectionName=Hereditary%20brachial%20plexopathy&search=Hereditary%20Neuropathy%20with%20liability%20to%20Pressure%20Palsies&topicRef=6207&anchor=H18&source=see_link#H18
- Burgunder, J. M., Schols, L., Baets, J., Andersen, P., Gasser, T., Szolnoki, Z., . . . Finsterer, J. (2011). EFNS guidelines for the molecular diagnosis of neurogenetic disorders: motoneuron, peripheral nerve and muscle disorders. Eur J Neurol, 18(2), 207-217. doi:10.1111/j.1468-1331.2010.03069.x
- Claritas_Genomics. (2021). PMP22 Deletion/Duplication. Retrieved from http://www.claritasgenomics.com/test/pmp22-deletionduplication/index.html
- CMTA. Genetic Testing. Retrieved from https://www.cmtausa.org/resource-center/treatment-management/genetic-testing/
- Cortese, A., Wilcox, J. E., Polke, J. M., Poh, R., Skorupinska, M., Rossor, A. M., . . . Reilly, M. M. (2020). Targeted next-generation sequencing panels in the diagnosis of Charcot-Marie-Tooth disease. Neurology, 94(1), e51-e61. doi:10.1212/wnl.0000000000008672
- Delague, V. (2013). Charcot-Marie-Tooth Neuropathy Type 4H. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK153601/
- DiVincenzo, C., Elzinga, C. D., Medeiros, A. C., Karbassi, I., Jones, J. R., Evans, M. C., . . . Higgins, J. J. (2014). The allelic spectrum of Charcot-Marie-Tooth disease in over 17,000 individuals with neuropathy. Mol Genet Genomic Med, 2(6), 522-529. doi:10.1002/mgg3.106
- Eichler, F. (2021). Hereditary sensory and autonomic neuropathies. Retrieved from https://www.uptodate.com/contents/hereditary-sensory-and-autonomic-neuropathies?search=Hereditary%20Neuropathy%20with%20liability%20to%20Pressure%20Palsies&topicRef=6207&source=see_link
- England, J. D., Gronseth, G. S., Franklin, G., Carter, G. T., Kinsella, L. J., Cohen, J. A., . . . Sumner, A. J. (2009). Practice Parameter: Evaluation of distal symmetric polyneuropathy: Role of laboratory and genetic testing (an evidence-based review). Neurology, 72(2), 185. doi:10.1212/01.wnl.0000336370.51010.a1
- FDA. (2021). Devices@FDA. Retrieved from https://www.accessdata.fda.gov/scripts/cdrh/devicesatfda/index.cfm
- GeneDx. (2018). Hereditary Neuropathy Panel. Retrieved from https://www.genedx.com/test-catalog/available-tests/hereditary-neuropathy-panel/
- Hamid Azzedine, E. L., and Mustafa A Salih. (2015). Charcot-Marie-Tooth Neuropathy Type 4C. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK1340/
- Invitae. (2020). Invitae Comprehensive Neuropathies Panel. Retrieved from https://www.invitae.com/en/physician/tests/03200/#info-panel-assay_information
- Kang, P. (2019). Overview of hereditary neuropathies. Retrieved from https://www.uptodate.com/contents/overview-of-hereditary-neuropathies?search=Hereditary%20Neuropathy%20with%20liability%20to%20Pressure%20Palsies&source=search_result&selectedTitle=1~16&usage_type=default&display_rank=1
- Kang, P. (2020a). Charcot-Marie-Tooth disease: Genetics, clinical features, and diagnosis. Retrieved from https://www.uptodate.com/contents/charcot-marie-tooth-disease-genetics-clinical-features-and-diagnosis
- Kang, P. (2020b). Charcot-Marie-Tooth disease: Management and prognosis. Retrieved from https://www.uptodate.com/contents/charcot-marie-tooth-disease-management-and-prognosis?topicRef=6220&source=see_link
- Kim, J.-W., Kim, Hee-Jin. (2013). Charcot-Marie-Tooth Neuropathy X Type 5. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK1876/
- Li, J. (2013). Charcot-Marie-Tooth Neuropathy Type 4J. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK169431/
- MNG_Laboratories. (2021). Charcot-Marie-Tooth Disease, Axonal (NGS Panel and Copy Number Analysis + mtDNA). Retrieved from https://mnglabs.com/tests/NGS345A/charcot-marie-tooth-disease-axonal-ngs-panel-and-copy-number-analysis-mtdna
- NINDS. (2007). Charcot-Marie-Tooth Disease Fact Sheet. Retrieved from https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Charcot-Marie-Tooth-Disease-Fact-Sheet#3092_5
- Pareyson, D., Saveri, P., & Pisciotta, C. (2017). New developments in Charcot-Marie-Tooth neuropathy and related diseases. Curr Opin Neurol, 30(5), 471-480. doi:10.1097/wco.0000000000000474
- Peter De Jonghe, M., PhD and Albena K Jordanova, PhD. (2011). Charcot-Marie-Tooth Neuropathy Type 2E/1F. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK1187/
- Prevention_Genetics. (2021). Charcot-Marie-Tooth (CMT) - Comprehensive Panel. Retrieved from https://www.preventiongenetics.com/testInfo?val=Charcot-Marie-Tooth+%28CMT%29+-+Comprehensive+Panel
- Rudnik-Schöneborn, S., Auer-Grumbach, M., & Senderek, J. (2020). Charcot-Marie-Tooth disease and hereditary motor neuropathies – Update 2020. Medizinische Genetik, 32(3), 207-219. doi:doi:10.1515/medgen-2020-2038
- Rudnik-Schoneborn, S., Tolle, D., Senderek, J., Eggermann, K., Elbracht, M., Kornak, U., . . . Zerres, K. (2016). Diagnostic algorithms in Charcot-Marie-Tooth neuropathies: experiences from a German genetic laboratory on the basis of 1206 index patients. Clin Genet, 89(1), 34-43. doi:10.1111/cge.12594
- Saporta, A. S., Sottile, S. L., Miller, L. J., Feely, S. M., Siskind, C. E., & Shy, M. E. (2011). Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann Neurol, 69(1), 22-33. doi:10.1002/ana.22166
- Schindler, A. (2014). TRPV4-Associated Disorders. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK201366/
- UTD. (2021). Patient education: Charcot-Marie-Tooth disease (The Basics). Retrieved from https://www.uptodate.com/contents/charcot-marie-tooth-disease-the-basics?search=Hereditary%20Neuropathy%20with%20liability%20to%20Pressure%20Palsies&topicRef=6207&source=see_link
- Vaeth, S., Christensen, R., Duno, M., Lildballe, D. L., Thorsen, K., Vissing, J., . . . Jensen, U. B. (2019). Genetic analysis of Charcot-Marie-Tooth disease in Denmark and the implementation of a next generation sequencing platform. Eur J Med Genet, 62(1), 1-8. doi:10.1016/j.ejmg.2018.04.003
- Züchner, S. (2013). Charcot-Marie-Tooth Neuropathy Type 2A. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK1511/
Coding Section
| Codes | Number | Description |
| CPT | 81324 | Pmp22 (peripheral myelin protein 22) (e.g., charcot-marie-tooth, hereditary neuropathy with liability to pressure palsies) gene analysis; duplication/deletion analysis |
| 81325 | Pmp22 (peripheral myelin protein 22) (e.g., charcot-marie-tooth, hereditary neuropathy with liability to pressure palsies) gene analysis; full sequence analysis | |
| 81326 | Pmp22 (peripheral myelin protein 22) (e.g., charcot-marie-tooth, hereditary neuropathy with liability to pressure palsies) gene analysis; known familial variant | |
| 81403 |
Mopath procedure level 4 Genes: GJB1 (gap junction protein, beta 1) (e.g., Charcot-Marie-Tooth X-linked), full gene sequence |
|
| 81404 |
Mopath procedure level 5 Genes: EGR2 (early growth response 2) (e.g., Charcot-Marie-Tooth), full gene sequence LITAF (lipopolysaccharide-induced TNF factor) (e.g., Charcot-Marie-Tooth), full gene sequence HSPB1 |
|
| 81405 |
Mopath procedure level 6 Genes: GDAP1 (ganglioside-induced differentiation-associated protein 1) (e.g., Charcot-Marie-Tooth disease), full gene sequence MPZ (myelin protein zero) (e.g., Charcot-Marie-Tooth), full gene sequence NEFL(neurofilament, light polypeptide) (e.g., Charcot-Marie-Tooth), full gene sequence RAB7A, PRX |
|
| 81406 |
Mopath procedure level 7 Genes: GARS (glycyl-tRNA synthetase) (e.g., Charcot-Marie-Tooth disease), full gene sequence MFN2 (mitofusin 2) (e.g., Charcot-Marie-Tooth disease), full gene sequence SH3TC2 (SH3 domain and tetratricopeptide repeats 2) (e.g., Charcot-Marie-Tooth disease), full gene sequence BSCL2, LMNA, FIG4 |
|
| 81448 (effective 1/1/2018) |
Hereditary peripheral neuropathies panel (e.g., Charcot-Marie-Tooth, spastic paraplegia), genomic sequence analysis panel, must include sequencing of at least 5 peripheral neuropathy-related genes (eg, BSCL2, GJB1, MFN2, MPZ, REEP1, SPAST, SPG11, and SPTLC1) |
|
| 96040 |
Medical genetics and genetic counseling services, each 30 minutes face-to-face with patient/family |
|
| S0265 |
Genetic counseling, under physician supervision, each 15 minutes |
|
| ICD-10-CM | G62.89 | Other specified polyneuropathies |
| M21.37 codes | Foot drop | |
|
M62.551-M62.579 codes |
Muscle wasting and atrophy, not elsewhere classified | |
| M62.81 | Muscle weakness (generalized) | |
| 009 codes | Supervision of High risk pregnancy | |
| R26.0, R26.2, R26.81, R26.89 | Abnormalities of gait and mobility | |
| Z13.71 |
Encounter for nonprocreative screening for genetic disease carrier status |
|
| Z13.79 | Encounter for other screening for genetic and chromosomal anomalies | |
| Z33.1 | Pregnancy state, incidental | |
| ICD-10-PCS (effective 10/01/15) |
Not applicable. ICD-10-PCS codes are only used for inpatient services. There are no ICD procedure codes for laboratory tests. |
|
| Type of Service | ||
| Place of Service |
Procedure and diagnosis codes on Medical Policy documents are included only as a general reference tool for each policy. They may not be all-inclusive.
This medical policy was developed through consideration of peer-reviewed medical literature generally recognized by the relevant medical community, U.S. FDA approval status, nationally accepted standards of medical practice and accepted standards of medical practice in this community, Blue Cross Blue Shield Association technology assessment program (TEC) and other nonaffiliated technology evaluation centers, reference to federal regulations, other plan medical policies, and accredited national guidelines.
"Current Procedural Terminology © American Medical Association. All Rights Reserved"
History From 2014 Forward
| 07/21/2022 | Annual review, policy rewritten for clarity, but, no change to policy intent. Also, updating description, rationale, and references. |
|
07/20/2021 |
Annual review, no change to policy intent. Updating coding, rationale and references. Removing regulatory status as that is now included in the rationale. |
|
07/21/2020 |
Annual review, no change to policy intent. Updating background, guidelines and references. |
|
07/22/2019 |
Annual review, rewording criteria #2 for clarity, adding medical necessity language related to hereditary motor neuropathy testing and adding a reimbursement statement.Updating coding. |
|
06/19/2019 |
Interim review. Genetic counseling is recommended is replacing Genetic counseling is Medically necessary. No other changes made |
|
11/15/2018 |
Corrected typo in policy section. No other changes. |
|
07/18/2018 |
Annual review, removing "ulnar/median" nerve in criteria #2, neutral to specific nerve. No other changes made. |
|
12/7/2017 |
Updating policy with 2018 coding. No other changes. |
|
06/23/2017 |
Interim review, updating background, description, policy, rationale, references and coding. |
|
04/25/2017 |
Updated category to Laboratory. No other changes. |
|
08/02/2016 |
Annual review, no change to policy intent. Updating background, description, rationale and references. |
|
08/19/2015 |
Annual review, no change to policy intent. Updated background, description, rationale and references. Added appendix 1, coding and guidelines. |
|
08/04/2014 |
Annual review. Updated description, background, rationale and references. No change to policy intent. |