
Prenatal Screening (Genetic) - CAM 358
Description
Prenatal screening encompasses any testing done to determine the health status of the pregnant individual and/or fetus. Genetic prenatal screening encompasses screening to determine risk of fetal abnormalities, including genetic and developmental abnormalities. Any individual undergoing screening tests, especially genetic carrier screenings, must realize the limitations of screening tests and the difference between screening and diagnostic testing. Screening refers to testing of asymptomatic or healthy individuals to search for a condition that may affect the pregnancy or individual, whereas diagnostic testing is used to either confirm or refute true abnormalities in an individual (Grant & Mohide, 1982; Lockwood & Magriples, 2020).
Regulatory Status
The FDA has approved many tests for conditions that can be included in a prenatal screening, such as HSV, chlamydia, gonorrhea, syphilis, and diabetes. Additionally, many labs have developed specific tests that they must validate and perform in house. These laboratory-developed tests (LDTs) are regulated by the Centers for Medicare & Medicaid (CMS) as high-complexity tests under the Clinical Laboratory Improvement Amendments of 1988 (CLIA ’88). LDTs are not approved or cleared by the U.S. Food and Drug Administration; however, FDA clearance or approval is not currently required for clinical use.
Policy
- For pregnant individuals and those persons seeking pre-conception care, any of the following testing* (See Note 1 below) of carrier status is considered MEDICALLY NECESSARY:
- Carrier screening for cystic fibrosis is in accordance with policy CAM 044 Genetic Testing for Cystic Fibrosis
- Carrier screening for Canavan disease, Tay-Sachs’ disease, familial dysautonomia, Gaucher disease, Fanconi Anemia, Niemann-Pick type A, Bloom syndrome and mucolipidosis IV
- Carrier screening for Fragile X syndrome
- Carrier screening for spinal muscular atrophy
- Carrier screening for hemoglobinopathies and/or thalassemia
- Carrier screening for hereditary hearing loss mutations (GJB2, GJB6, and other hereditary hearing loss-related mutations) in parents according to the policy CAM 294 Genetic Testing for Hereditary Hearing Loss
- Carrier screening for other genetic disorders when there is a family history of a genetic disorder and a properly validated test is available. When there is a known familial mutation, testing should be limited to that mutation, when possible. (See General Genetic Testing policy for more details on appropriate criteria for genetic testing.)
- Next generation sequencing (NGS) panel testing, as long as a single appropriate AMA genetic sequencing procedure test code is submitted.
- Carrier screening* (See Note 1 below) of the biological father is considered MEDICALLY NECESSARY when the biological mother is known or found to be a carrier of a recessively inherited disorder. Carrier testing limitation:
- Repeat carrier screening for the same disorder is considered NOT MEDICALLY NECESSARY.
- Fetal RHD genotyping using maternal plasma is considered MEDICALLY NECESSARY in RHD negative pregnant individuals.
- Pre-conception carrier screening in patients with a family history of a known inherited disorder and if positive, testing of the partner is considered MEDICALLY NECESSARY.
- Prenatal genetic testing of a fetus is considered MEDICALLY NECESSARY if high risk for genetic disorder.
- Carrier screening more than once per lifetime is considered NOT MEDICALLY NECESSARY.
The following does not meet coverage criteria due to a lack of available published scientific literature confirming that the test(s) is/are required and beneficial for the diagnosis and treatment of a patient’s illness.
- Pre-conceptional or prenatal genetic testing for inherited medical disorders that do not meet the above criteria is NOT MEDICALLY NECESSARY.
Note 1: Carrier screening should be performed using the most appropriate carrier test (e.g., dosage/deletion for SMN1 and NOT full gene sequencing; DMD del/dup testing and NOT full gene sequencing).
Rationale
Prenatal screening is a part of overall prenatal care to promote optimal care of both mother and baby. Prenatal screening allows for assessment and monitoring of the fetus for the presence of congenital defects or disease. Various professional medical organizations provide guidelines for prenatal screening. “Screening is an offer on the initiative of the health system or society, rather than a medical intervention in answer to a patient’s complaint or health problem. Screening aims at obtaining population health gains through early detection that enables prevention or treatment” (de Jong et al., 2015).
Genetic screening tests, including carrier screening for genetic mutations and fetal testing for chromosomal aneuploidy, can be a part of prenatal screening. Aneuploidy screening may be performed on cell-free DNA in maternal circulation or by examining maternal serum levels of specific biochemical markers for trisomy (Lockwood & Magriples, 2020). These non-invasive prenatal testing (NIPT) can possibly decrease the number of more invasive procedures and the risks of unwanted side effects. A chromosomal microarray (CMA) can screen all chromosomes in a single test and “can detect many very small variants that cannot be detected by traditional karyotyping” (de Jong et al., 2015). The American College of Obstetricians and Gynecologists (ACOG) recommends CMA for instances where the ultrasound of a fetus shows a major structural abnormality (ACOG, 2016a). CMA in this situation should be performed on DNA from amniotic fluid, chorionic villus cells, or cord blood, rather than on maternal serum cell-free DNA since the process does not include an amplification step and the maternal DNA signal would be many times higher than the fetal DNA (Miller, 2020).
Several companies, such as LabCorp, have developed panels to test for potential genetic mutations in pregnant individuals, or in individuals planning to become pregnant. This includes the Inheritest® Carrier Screening which encompasses six different panels to identify potential genetic mutations. These six panels include the Inheritest® 500 PLUS Panel (which screens 525 genes for several clinically relevant genetic disorders), the Inheritest® Comprehensive Panel (which screens for more than 110 disorders), the Inheritest® Ashkenazi Jewish Panel (which screens for more than 40 Ashkenazi Jewish related disorders), the Inheritest® Society-Guided Panel (which screens for more than 13 disorders highlighted in the American College of Medical Genetics and Genomics and the American Congress of Obstetricians and Gynecologists guidelines), the Inheritest® Core Panel (which screens for cystic fibrosis, fragile X syndrome, and spinal muscular atrophy), and the Inheritest® CF/SMA (spinal muscular atrophy) Panel (which screens only for cystic fibrosis and spinal muscular atrophy) (LabCorp, 2020).
Red blood cell antigen discrepancy between a mother and fetus may also occur during pregnancy. This is known as hemolytic disease of the fetus and newborn (HDFN), and causes maternal antibodies to destroy the red blood cells of the neonate or fetus (Calhoun, 2020). Alloimmunization is the immune response which occurs in the mother due to foreign antigens after exposure to genetically foreign cells, occurring almost exclusively in mothers with type O blood. However, while ABO blood type incompatibility is identified in almost 15% of pregnancies, HDFN is only identified in approximately 4% of pregnancies (Calhoun, 2020). Another important inherited antigen sometimes found on the surface of red blood cells is known as the Rhesus (Rh)D antigen. During pregnancy and delivery, individuals who are RhD negative may be exposed to RhD positive fetal cells, which can lead to the development of anti-RhD antibodies. This exposure typically happens during delivery and affects subsequent pregnancies; infants with RhD incompatibility tend to experience a more severe form of HDFN than those with ABO incompatibility. The clinical presentation of HDFN may be mild (such as hyperbilirubinemia with mild to moderate anemia) to severe and life-threatening anemia (such as hydrops fetalis). Less severely affected infants may develop hyperbilirubinemia within the first day of life; infants with RhD HDFN may also present with symptomatic anemia requiring a blood transfusion. In more severe cases, infants with severe life-threatening anemia, such as hydrops fetalis, may exhibit shock at delivery requiring an emergent blood transfusion (Calhoun, 2020).
The administration of anti-D immune globulin has been able to dramatically reduce, but not eliminate, the number of RhD alloimmunization cases. “Anti-D immune globulin is manufactured from pooled plasma selected for high titers of IgG antibodies to D-positive erythrocytes” (Moise, 2020). Before the development of this anti-D immune globulin, it has been reported that 16% of pregnant RhD-negative individuals with two deliveries of RhD-positive ABO-compatible infants became alloimmunized. However, this rate falls to 1-2% with routine postpartum administration of a single dose of anti-D immune globulin. An additional administration in the third trimester of pregnancy further reduces the incidents of alloimmunization to 0.1 – 0.3% (Moise, 2020).
Fetal RhD genotyping using cell-free fetal DNA from maternal plasma can be performed to identify fetal blood type most accurately after 11 weeks of gestation. While the United States has not implemented fetal RhD genotyping for routine prophylaxis and fetal monitoring protocols, several European countries, such as Denmark, the Netherlands, England, Sweden, France and Finland, do utilize fetal RhD determination so that the administration of anti-D immune globulin can be avoided when an RhD-negative fetus is identified (Moise, 2020). Daniels et al. (2007) report that approximately 40% of RhD-negative pregnant individuals are carrying a RhD-negative fetus; genotypic screening would, therefore, be very valuable in preventing these individuals from receiving unnecessary anti-D immune globulin. Kent et al. (2014) suggest that the administration of anti-D immune globulin to the one third of pregnant individuals who do not require this administration is unethical, and that the availability of RhD genotyping to all RhD-negative pregnant individuals would assist in more informed choices being made regarding anti-D immune globulin administration. Finning et al. (2008) agree with the previous statements, declaring that “high throughput RHD genotyping of fetuses in all RhD negative [individuals] is feasible and would substantially reduce unnecessary administration of anti-RhD immunoglobulin to RhD negative pregnant [individuals] with an RhD negative fetus.”
Analytical Validity
A prospective cohort study by de Haas et al. (2016) completed a nationwide program in the Netherlands to determine the sensitivity of fetal RhD screening for the safe guidance of targeted anti-immune globulin prophylaxis. A total of 25,789 RhD-negative pregnant individuals participated in this study. Fetal testing for the RHD gene was assessed in the 27th week of pregnancy. Fetal RHD test results were compared to serological cord blood results after birth. “Sensitivity for detection of fetal RHD was 99.94% (95% confidence interval 99.89% to 99.97%) and specificity was 97.74% (97.43% to 98.02%). Nine false-negative results for fetal RHD testing were registered (0.03%, 95% confidence interval 0.01% to 0.06%)” (de Haas et al., 2016). They conclude that fetal RhD testing is a highly reliable testing method.
Manfroi et al. (2018) completed fetal RhD genotyping with real-time polymerase chain reaction (qPCR) using cell-free fetal DNA extracted from maternal plasma. A commercial multiple-exon assay was used to determine fetal RHD genotypic accuracy. A total of 367 plasma samples obtained between the 24th and 28th weeks of pregnancy were used for this study. Neonatal results were available for 284 of the pregnancies. The sensitivity was reported at 100% and specificity at 97.5%. The diagnostic accuracy was 96.1% with the inclusion of 9/284 inconclusive results (Manfroi et al., 2018). The authors conclude that this is therefore an accurate and reliable tool for targeted prenatal immunoprophylaxis.
Liang et al. (2019) used cell-free plasma DNA to assess the clinical utility of using an expanded noninvasive prenatal screening (“NIPS-Plus”) to detect aneuploidy and genome-wide microdeletion/microduplication syndromes (MMS). Of the 94,085 individuals with singleton pregnancies enrolled in the study, 1128 were suspected of having clinically significant fetal chromosomal abnormalities. Follow-up testing in the study reported the positive predictive values (PPVs) of 95%, 82%, 46%, 29%, and 47% for T21, T18, T13, rare trisomies, and sex chromosome aneuploidies, respectively. For known MMS (n = 32), PPVs were 93% (DiGeorge), 68% (22q11.22 microduplication), 75% (Prader-Willi/Angelman), and 50% (Cri du Chat). Thus, the researchers conclude that “the data have potential significance in demonstrating the usefulness of cfDNA profiling” and that “NIPS-Plus can be used as a first-tier pregnancy screening method to improve detection rates of clinically significant fetal chromosome abnormalities” (Liang et al., 2019).
Clinical Utility and Validity
Education and counseling are a key factor in prenatal screening and diagnostic tests. Yesilcinar and Guvenc (2021) found that a proactive intervention approach decreased anxiety and decisional conflict in the pregnant individual and increased attitudes towards the tests, having a positive effect on the pregnant individual’s knowledge level and decision satisfaction. This allowed the individual to make more informed decisions, such as opting to have screening and diagnostic testing performed. Decreasing anxiety during pregnancy is beneficial to the fetus and individuals receiving educational intervention showed decreased anxiety when receiving genetic screening results as compared to individuals not receiving the same intervention (Yesilcinar & Guvenc, 2021). Migliorini et al. (2020) have also reported that the use of cell free DNA (cfDNA) screening, combined with a detailed ultrasound examination, as a first-trimester risk assessment is associated with improved maternal reassurance and satisfaction and decreased anxiety, as compared to individuals who received standard first-trimester combined screening with nuchal translucency (NT) and biochemistry (Migliorini et al., 2020).
Biro et al. (2020) report on a noninvasive prenatal testing method for congenital heart disease, utilizing the measurement of cell-free nucleic acid and protein biomarkers in maternal blood. Congenital heart disease is considered the most common fetal malformation. While prenatal ultrasonography is currently used to diagnose congenital heart disease, it is not the most accurate method. After a large review completed with PubMed and Web of Sciences databases, the authors conclude that most fetal congenital heart disease related disorders can be diagnosed by noninvasive prenatal testing (NIPT) techniques. Further, cell-free RNAs and circulating proteins are potential biomarkers for fetal congenital heart disease and may be able to improve the detection rate in early pregnancies (Biro et al., 2020).
A study by Persico et al. (2016) investigated the clinical implication of cfDNA testing in high-risk pregnancies. In their cohort of 259 singleton pregnancies, cfDNA testing provided results in 249 (96.1%). Further, cfDNA testing identified 97.2% (35/36) of trisomy 21, 100% (13/13) of trisomy 18, 100% of trisomy 13 (5/5), and 75% of sex chromosome aneuploidies (3/4). The authors conclude that “a policy of performing an invasive test in [individuals] with a combined risk of ≥ 1 in 10 or NT ≥ 4 mm and offering cfDNA testing to the remaining cases would detect all cases of trisomy 21, 18 or 13, 80% of sex aneuploidies and 62.5% of other defects and would avoid an invasive procedure in 82.4% of euploid fetuses” (Persico et al., 2016). These data support the earlier meta-analysis that reported NIPT sensitivity of trisomy 21, trisomy 18, and trisomy 13 of 99%, 96.8%, and 92.1%, respectively and specificities of 99.92%, 99.85%, and 99.80%, respectively, for trisomies 21, 18, and 13 (Dondorp et al., 2015; Gil et al., 2014).
A multi-year study of more than 5000 patients in public hospitals in Spain examined the effect of NIPT on the number of invasive procedures performed, showing that the introduction of NIPT drastically reduced the incidences of invasive procedures. The data shows that despite a 60.5% reduction occurred in invasive procedures, the chromosomopathy detection rate was unaffected; moreover, the ratio of positive invasive procedures was improved to 50%, indicating that unwarranted invasive procedures had been avoided (Martinez-Payo et al., 2018). The authors of the study concluded, “NIPT introduction has caused a significant reduction of 60.5% of IP [invasive procedures] in high chromosomopathy risk patients after combined screening without modifying detection rate” (Martinez-Payo et al., 2018).
A meta-analysis was completed by Mackie et al. (2017), researching the accuracy of cell-free fetal DNA NIPT testing in singleton pregnancies. A total of 117 studies were included, analyzing 18 different conditions. For RHD testing, a sensitivity of 0.993 and specificity of 0.984 was identified and for fetal sex identification, a sensitivity of 0.989 and a specificity of 0.996 was calculated (Mackie et al., 2017). With such high sensitivity and specificity calculations, NIPT testing for fetal sex and RHD status may be considered accurate diagnostic tools.
Clausen et al. (2014) completed a two-year evaluation of nationwide prenatal RhD screening in Denmark. A total of 12,668 pregnancies were analyzed, with blood samples drawn in week 25 of pregnancy. DNA was extracted from these blood samples and was analyzed for the RHD gene. Results were later compared to the serological typing of the newborns after birth. “The sensitivity for the detection of fetal RHD was 99.9% (95% CI: 99.7 – 99.9%). Unnecessary recommendation of prenatal RhD prophylaxis was avoided in 97.3% of the [individuals] carrying an RhD-negative fetus. Fetuses that were seropositive for RhD were not detected in 11 pregnancies (0.087%)” (Clausen et al., 2014). This study shows high sensitivity of fetal RHD genotyping, results which were recently supported by another large-scale meta-analysis completed by Yang et al. (2019), focusing on NIPT testing for fetal RhD status. A total of 3921 results confirmed that “High-throughput NIPT is sufficiently accurate to detect fetal RhD status in RhD-negative [individuals] and would considerably reduce unnecessary treatment with routine anti-D immunoglobulin” (Yang et al., 2019).
Darlington et al. (2018) completed an analysis of 11 French Obstetric Departments with a total of 949 patients to determine the effectiveness of RhD genotyping. The patients were separated into two groups (genotyping group: n = 515, and control group: n = 335). The authors concluded that “Early knowledge of the RHD status of the fetus using non-invasive fetal RHD genotyping significantly improved the management of RHD negative pregnancies with a small increase in cost” (Darlington et al., 2018).
Runkel et al. (2020) completed a systematic review to determine the benefit of NIPT for fetal RhD status in RhD-negative pregnant individuals because “All non-sensitized Rhesus D (RhD)-negative pregnant [individuals] in Germany receive antenatal anti-D prophylaxis without knowledge of fetal RhD status.” The meta-analysis included data from 60,000 participants, with the focus of the research on the impact of fetal and maternal morbidity. The researchers concluded that “NIPT for fetal RhD status is equivalent to conventional serologic testing using the newborn’s blood. Studies investigating patient-relevant outcomes are still lacking” (Runkel et al., 2020).
It is notable that the field continues to evolve, with potential shifts from one testing method to another in pursuit of optimality and comprehensiveness. A multicenter retrospective study of singleton high-risk pregnancies for chromosomal abnormalities was conducted by Zhu et al. (2020) to evaluate the utility of expanded noninvasive prenatal screening as compared with chromosomal microarray analysis (CMA). The analysis enrolled subjects who underwent expanded NIPS and CMA sequentially during pregnancy from 2015 through 2019. The study demonstrated that of the 943 high‐risk pregnancies, 550 (58.3%) cases had positive NIPS results, while positive CMA results were detected in 308 (32.7%) cases, and the agreement rates between NIPS and CMA were 82.3%, 59.6% and 25.0% for trisomy 21, 18 and 13, respectively. Regarding rare aneuploidies and segmental imbalances, NIPS and CMA results were concordant in 7.5% and 33.3% of cases. However, copy number variants were better detected with CMA than with NIPS and additional genetic aberrations were detected by CMA in one of 17 high-risk pregnancies that were otherwise passed over when processed with NIPS. The researchers contend that CMA should be offered for high‐risk pregnancies to provide comprehensive detection of chromosomal abnormalities in these pregnancies (Zhu et al., 2020).
This policy focuses on laboratory testing performed during pre-conception and/or prenatal periods as part of a comprehensive prenatal care program.
American College of Obstetricians and Gynecologists (ACOG)
ACOG has several practice guidelines related to prenatal care as well as both pre-conception and prenatal testing. ACOG recommendations and guidelines include the following:
- Genetic Testing and Genetic Counseling: Concerning genetic testing and genetic counseling, ACOG recommends:
- “A hereditary cancer risk assessment is the key to identifying patients and families who may be at increased risk of developing certain types of cancer. Assessments should be performed by obstetrician–gynecologists or other obstetric–gynecologic providers and should be updated regularly. ... If a hereditary cancer risk assessment suggests an increased risk of a hereditary cancer syndrome, referral to a specialist in cancer genetics or a health care provider with expertise in genetics is recommended for expanded gathering of family history information, risk assessment, education, and counseling, which may lead to genetic testing and tailored cancer screening or risk reduction measures, or both.” (ACOG, 2019)
- “The routine use of whole-genome or whole-exome sequencing for prenatal diagnosis is not recommended outside of the context of clinical trials until sufficient peer-reviewed data and validation studies are published.” This was reaffirmed in 2020 (ACOG, 2016a).
- Chromosomal microarray analysis (CMA) is recommended for patients with a fetus with at least one major structure abnormality identified via ultrasound. CMA can be considered for all pregnant [individuals] who undergo prenatal diagnostic testing; however, “In a patient with a structurally normal fetus who is undergoing invasive prenatal diagnostic testing, either fetal karyotyping or a chromosomal microarray analysis can be performed. Chromosomal microarray analysis of fetal tissue (i.e., amniotic fluid, placenta, or products of conception) is recommended in the evaluation of intrauterine fetal death or stillbirth when further cytogenetic analysis is desired because of the test’s increased likelihood of obtaining results and improved detection of causative abnormalities” (ACOG, 2016a). This was reaffirmed in 2020.
- “All patients who are considering pregnancy or are already pregnant, regardless of screening strategy and ethnicity, should be offered carrier screening for cystic fibrosis and spinal muscular atrophy, as well as a complete blood count and screening for thalassemias and hemoglobinopathies. Fragile X premutation carrier screening is recommended for [individuals] with a family history of fragile X-related disorders or intellectual disability suggestive of fragile X syndrome, or [individuals] with a personal history of ovarian insufficiency. Additional screening also may be indicated based on family history or specific ethnicity” (Romero et al., 2017). This was reaffirmed in 2020 (ACOG, 2020).
- “The American College of Obstetricians and Gynecologists discourages direct-to-consumer genetic testing without appropriate counseling. ... Patients may present after direct-to-consumer testing already has been performed, and clinicians should be prepared to review these results or refer to a health care professional with the appropriate knowledge, training, and experience in interpreting test results. ... Patients may present after direct-to-consumer testing already has been performed, and clinicians should be prepared to review these results or refer to a health care professional with the appropriate knowledge, training, and experience in interpreting test results. ... Given the insufficient data to support the use of single nucleotide polymorphisms (SNP) testing for medical purposes, SNP testing to provide individual risk assessment for a variety of diseases or to tailor drug therapy outside of an institutional review board-approved research protocol is not recommended. The American College of Obstetricians and Gynecologists recommends that the use of these technologies be viewed as investigational at this time” (ACOG, 2021).
- ACOG “recommends considering whole-exome sequencing when specific genetic tests available for a phenotype, including targeted sequencing tests, have failed to arrive at a diagnosis in a fetus with multiple congenital anomalies suggestive of a genetic disorder” (Vora et al., 2018); however, they note that “Cascade testing has been shown to be cost effective in part because testing for specific mutations (eg, those identified in the affected relative) is less expensive than whole-gene sequencing.” This was reaffirmed in 2020 (Witkop & ACOG, 2018).
- Prenatal Diagnostic Testing for Genetic Disorders: Concerning prenatal diagnostic testing for genetic disorders, ACOG has published the following recommendations (ACOG, 2016b):
- “An abnormal FISH result should not be considered diagnostic. Therefore, clinical decision making based on information from FISH should include at least one of the following additional results: confirmatory traditional metaphase chromosome analysis or chromosomal microarray, or consistent clinical information
- Prenatal genetic testing cannot identify all abnormalities or problems in a fetus, and any testing should be focused on the individual patient’s risks, reproductive goals and preferences
- Genetic testing should be discussed as early as possible in pregnancy, ideally at the first obstetric visit, so that first-trimester options are available” (ACOG, 2016b). This guideline was reaffirmed in 2021.
- Prevention of Rh D Alloimmunization: Concerning the prevention of Rh D alloimmunization, ACOG has published the guidelines supporting the administration of anti-D immune globulin to individuals in various scenarios. However, these guidelines do not mention the use of cell-free fetal DNA for fetal RHD testing to determine if anti-D immune globulin is needed (ACOG, 2017b).
- Genetic Carrier Screening: Concerning genetic carrier screening, including testing for specific conditions, ACOG recommends [(Rink et al., 2017; Romero et al., 2017) reaffirmed 2020]:
- “Carrier screening and counseling ideally should be performed before pregnancy.”
- “If an individual is found to be a carrier for a specific condition, the individual’s reproductive partner should be offered testing in order to receive informed genetic counseling about potential reproductive outcomes. Concurrent screening of the patient and her partner is suggested if there are time constraints for decisions about prenatal diagnostic evaluation.”
- “Carrier screening for a particular condition generally should be performed only once in a person’s lifetime, and the results should be documented in the patient’s health record. Because of the rapid evolution of genetic testing, additional mutations may be included in newer screening panels. The decision to rescreen a patient should be undertaken only with the guidance of a genetics professional who can best assess the incremental benefit of repeat testing for additional mutations.”
- “Prenatal carrier screening does not replace newborn screening, nor does newborn screening replace the potential value of prenatal carrier screening.”
- "The cost of carrier screening for an individual condition may be higher than the cost of testing through commercially available expanded carrier screening panels. When selecting a carrier screening approach, the cost of each option to the patient and the health care system should be considered.”
- “Screening for spinal muscular atrophy should be offered to all [individuals] who are considering pregnancy or are currently pregnant. In patients with a family history of spinal muscular atrophy, molecular testing reports of the affected individual and carrier testing of the related parent should be reviewed, if possible, before testing. If the reports are not available, SMN1 deletion testing should be recommended for the low-risk partner.”
- “Cystic fibrosis carrier screening should be offered to all [individuals] who are considering pregnancy or are currently pregnant. Complete analysis of the CFTR gene by DNA sequencing is not appropriate for routine carrier screening.”
- “A complete blood count with red blood cell indices should be performed in all [individuals] who are currently pregnant to assess not only their risk of anemia but also to allow assessment for risk of a hemoglobinopathy. Ideally, this testing also should be offered to [individuals] before pregnancy. A hemoglobin electrophoresis should be performed in addition to a complete blood count if there is suspicion of hemoglobinopathy based on ethnicity (African, Mediterranean, Middle Eastern, Southeast Asian, or West Indian descent). If red blood cell indices indicate a low mean corpuscular hemoglobin or mean corpuscular volume, hemoglobin electrophoresis also should be performed.”
- “Fragile X premutation carrier screening is recommended for [individuals] with a family history of fragile X-related disorders or intellectual disability suggestive of fragile X syndrome and who are considering pregnancy or are currently pregnant.”
- “If a [individual] has unexplained ovarian insufficiency or failure or an elevated follicle-stimulating hormone level before age 40 years, fragile X carrier screening is recommended to determine whether she has an FMR1 premutation.”
- “All identified individuals with intermediate results and carriers of a fragile X premutation or full mutation should be provided follow-up genetic counseling to discuss the risk to their offspring of inheriting an expanded full-mutation fragile X allele and to discuss fragile X-associated disorders (premature ovarian insufficiency and fragile X tremor/ataxia syndrome).”
- “Prenatal diagnostic testing for fragile X syndrome should be offered to known carriers of the fragile X premutation or full mutation.”
- “DNA-based molecular analysis (e.g., Southern blot analysis and polymerase chain reaction) is the preferred method of diagnosis of fragile X syndrome and of determining FMR1 triplet repeat number (e.g., premutations). In rare cases, the size of the triplet repeat and the methylation status do not correlate, which makes it difficult to predict the clinical phenotype. In cases of this discordance, the patient should be referred to a genetics professional.”
- “When only one partner is of Ashkenazi Jewish descent, that individual should be offered screening first. If it is determined that this individual is a carrier, the other partner should be offered screening. However, the couple should be informed that the carrier frequency and the detection rate in non-Jewish individuals are unknown for most of these disorders, except for Tay.Sachs disease and cystic fibrosis. Therefore, it is difficult to accurately predict the couple’s risk of having a child with the disorder.”
- “Screening for Tay-Sachs disease should be offered when considering pregnancy or during pregnancy if either member of a couple is of Ashkenazi Jewish, French-Canadian, or Cajun descent. Those with a family history consistent with Tay-Sachs disease also should be offered screening. When one member of a couple is at high risk (i.e., of Ashkenazi Jewish, French-Canadian, or Cajun descent or has a family history consistent with Tay-Sachs disease) but the other partner is not, the high-risk partner should be offered screening. If the high-risk partner is found to be a carrier, the other partner also should be offered screening. Enzyme testing in pregnant [individuals] and [individuals] taking oral contraceptives should be performed using leukocyte testing because serum testing is associated with an increased false-positive rate in these populations. If Tay-Sachs disease screening is performed as part of pan-ethnic expanded carrier screening, it is important to recognize the limitations of the mutations screened in detecting carriers in the general population. In the presence of a family history of Tay-Sachs disease, expanded carrier screening panels are not the best approach to screening unless the familial mutation is included on the panel” (Rink et al., 2017).
- Regarding expanded carrier screening panels, ACOG recommends that “the disorders selected for inclusion should meet several of the following consensus-determined criteria: have a carrier frequency of 1 in 100 or greater, have a well-defined phenotype, have a detrimental effect on quality of life, cause cognitive or physical impairment, require surgical or medical intervention, or have an onset early in life.” ACOG further states that “screened conditions should be able to be diagnosed prenatally and may afford opportunities for antenatal intervention to improve perinatal outcomes, changes to delivery management to optimize newborn and infant outcomes, and education of the parents about special care needs after birth” (Romero et al., 2017).
- Carrier Screening in the Age of Genomic Medicine: Concerning carrier screening in the age of genomic medicine, the ACOG has published the following guidelines (ACOG, 2017a):
- “Ethnic-specific, panethnic and expanded carrier screening are acceptable strategies for prepregnancy and prenatal carrier screening
- If a patient requests a screening strategy other than the one used by the obstetrician-gynecologist or other health care provider, the requested test should be made available to her after counseling on its limitations, benefits, and alternatives
- All patients who are considering pregnancy or already pregnant, regardless of screening strategy and ethnicity, should be offered carrier screening for cystic fibrosis and spinal muscular atrophy, as well as a complete blood count and screening for thalassemias and hemoglobinopathies. Fragile X premutation carrier screening is also recommended for [individuals] with a family history of fragile x-related disorders or intellectual disability suggestive of fragile X syndrome, or [individuals] with a personal history of ovarian insufficiency. Additional screening also may be indicated based on family history or specific ethnicity
- If a [individual] is found to be a carrier for a specific condition, her reproductive partner should be offered screening to provide accurate genetic counseling for the couple with regard to the risk of having an affected child. Additional genetic counseling should be provided to discuss the specific condition, residual risk, and options for prenatal testing.
- Individuals with a family history of a genetic disorder may benefit from the identification of the specific familial mutation or mutations rather than carrier screening. Knowledge of the specific familial mutation may allow for more specific and rapid prenatal diagnosis.
- Given the multitude of conditions that can be included in expanded carrier screening panels, the disorders selected for inclusion should meet several of the following consensus-determined criteria: have a carrier frequency of 1 in 100 or greater, have a well-defined phenotype, have a detrimental effect on quality of life, cause cognitive or physical impairment, require surgical or medical intervention, or have an onset early in life. Additionally, screened conditions should be able to be diagnosed prenatally and may afford opportunities for antenatal intervention to improve perinatal outcomes, changes to delivery management to optimize newborn and infant outcomes, and education of the parents about special care needs after birth.
- Carrier screening panels should not include conditions primarily associated with a disease of adult onset” (ACOG, 2017a). This guideline was reaffirmed in 2020
American College of Medical Genetics and Genomics (ACMG)
The American College of Medical Genetics and Genomics (ACMG) recommends that the following (Gregg et al., 2016):
- “Allowing patients to select diagnostic or screening approaches for the detection of fetal aneuploidy and/or genomic changes that are consistent with their personal goals and preferences.”
- “Informing all pregnant [individuals] that diagnostic testing (CVS or amniocentesis) is an option for the detection of chromosome abnormalities and clinically significant CNVs [copy-number variants].”
- “Informing all pregnant [individuals] that NIPS [non-invasive prenatal screening] is the most sensitive screening option for traditionally screened aneuploidies (i.e., Patau, Edwards, and Down syndromes).”
- “Offering diagnostic testing when a positive screening test result is reported after NIPS.”
- The ACMG does NOT recommend “NIPS to screen for autosomal aneuploidies other than those involving chromosomes 13, 18, and 21.”
- "Offering diagnostic testing for a no-call NIPS result due to low fetal fraction if maternal blood for NIPS was drawn at an appropriate gestational age. A repeat blood draw is NOT appropriate.”
- “Offering aneuploidy screening other than NIPS in cases of significant obesity.”
- “Offering diagnostic testing when a positive screening test result is reported after screening for sex chromosome aneuploidies.”
- “Offering diagnostic testing (CVS or amniocentesis) with CMA when NIPS identifies a CNV.”
- ACMG does NOT recommend “NIPS to screen for genome-wide CNVs. If this level of information is desired, then diagnostic testing (e.g., chorionic villous sampling or amniocentesis) followed by CMA is recommended.”
- “Offering aneuploidy screening other than NIPS for patients with a history of bone marrow or organ transplantation from a male donor or donor of uncertain biologic sex.”
In 2021, ACMG released an updated guideline for screening for autosomal recessive and X-linked conditions during pregnancy and preconception. Their practice resource reviews aim to recommend “a consistent and equitable approach for offering carrier screening to all individuals during pregnancy and preconception” and replaces any earlier ACMG position statements on prenatal/preconception expanded carrier screening and provide the following recommendations:
- “Analytical validity of carrier screening is to be established by a laboratory in compliance with CLIA/CAP regulations and adhering to ACMG Laboratory Standards and Guidelines.”
- “As evidence evolves, ClinVar and ClinGen continually update pathogenicity of variants and the association between genes and conditions, respectively.”
- “Carrier screening enables those screened to consider their reproductive risks, reproductive options, and to make informed decisions.”
- “Published evidence supports clinical utility for carrier screening of multiple conditions simultaneously.”
- “The phrase “expanded carrier screening” be replaced by “carrier screening”.”
- “Adopting a more precise tiered system based on carrier frequency:
- Tier 4: < 1/200 carrier frequency (includes Tier 3) genes/condition will vary by lab
- Tier 3: ≥ 1/200 carrier frequency (includes Tier 2) includes X-linked conditions
- Tier 2: ≥ 1/100 carrier frequency (includes Tier 1)
- Tier 1: CF [Cystic Fibrosis] + SMA [spinal muscular atrophy] + Risk Based Screening
- “Tier 1 screening conveys the recommendations previously adopted by ACMG and ACOG” and “adopts an ethnic and population neutral approach when screening for cystic fibrosis and spinal muscular atrophy. Beyond these two conditions, additional carrier screening is determined after risk assessment, which incorporates personal medical and family history as well as laboratory and imaging information where appropriate.”
- “Tier 2 carrier screening stems from an ACOG recommendation for conditions that have a severe or moderate phenotype and a carrier frequency of at least 1/100.” However, “data demonstrate that carrier screening for two common conditions using a carrier frequency threshold of 1/100 may not be equitable across diverse populations. Others have shown that limiting the carrier frequency to ≥ 1/100 creates missed opportunities to identify couples at risk for serious conditions.”
- “We define Tier 3 screening as carrier screening for conditions with a carrier frequency ≥ 1/200 ... Tier 2 and Tier 3 screening prioritize carrier frequency as a way to think about conditions most appropriate for screening in the general population. However, when ACOG proposed this level, they did not specify whether it was thinking about carrier frequency in terms of the global population or subpopulations. We use “carrier frequency” to mean in any ethnic group with reasonable representation in the United States.”
- “Tier 4 includes genes less common than those in Tier 3 and can identify additional at-risk couples. Tier 4 has no lower limit carrier screening frequency and can greatly extend the number of conditions screened ... the clinical validity at this level of carrier screening may be less compelling, therefore we suggest reserving this level of screening for consanguineous pregnancies (second cousins or closer) and in couples where family or medical history suggests Tier 4 screening might be beneficial ... Importantly, patients should understand that their chance of being a carrier for one or more conditions increases as the number of conditions screened is increased.”
- “All pregnant patients and those planning a pregnancy should be offered Tier 3 carrier screening.
- Tier 4 screening should be considered:
- When a pregnancy stems from a known or possible consanguineous relationship (second cousins or closer);
- When a family or personal medical history warrants.
- ACMG does NOT recommend:
- Offering Tier 1 and/or Tier 2 screening, because these do not provide equitable evaluation of all racial/ethnic groups.
- Routine offering of Tier 4 panels.
- “Carrier screening paradigms should be ethnic and population neutral and more inclusive of diverse populations to promote equity and inclusion.”
- “All pregnant patients and those planning a pregnancy should be offered Tier 3 carrier screening for autosomal recessive (Tables 1 – 5) and X-linked (Table 6) conditions.”
- “Reproductive partners of pregnant patients and those planning a pregnancy may be offered Tier 3 carrier screening for autosomal recessive conditions (Tables 1 – 5) when carrier screening is performed simultaneously with their partner.”
- “All XX patients should be offered screening for only those X-linked genes listed in Table 6 as part of Tier 3 screening.”
- “When Tier 1 or Tier 2 carrier screening was performed in a prior pregnancy, Tier 3 screening should be offered” (Gregg et al., 2021).
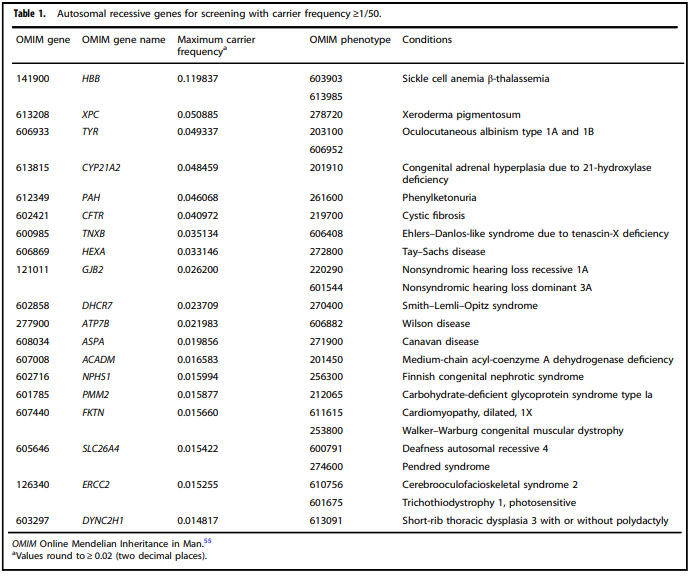
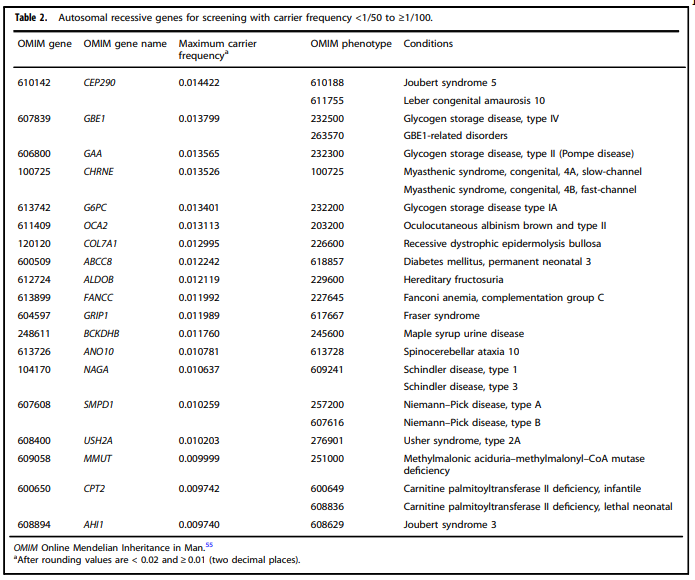
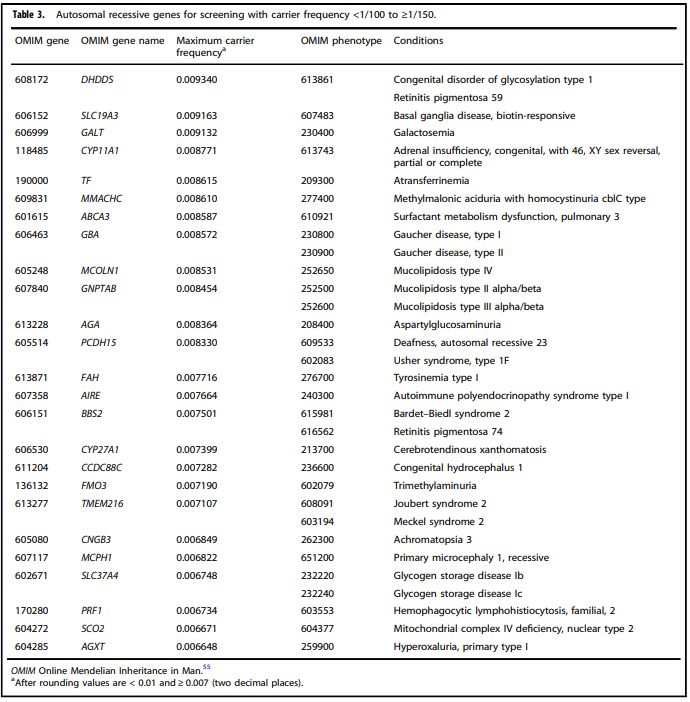
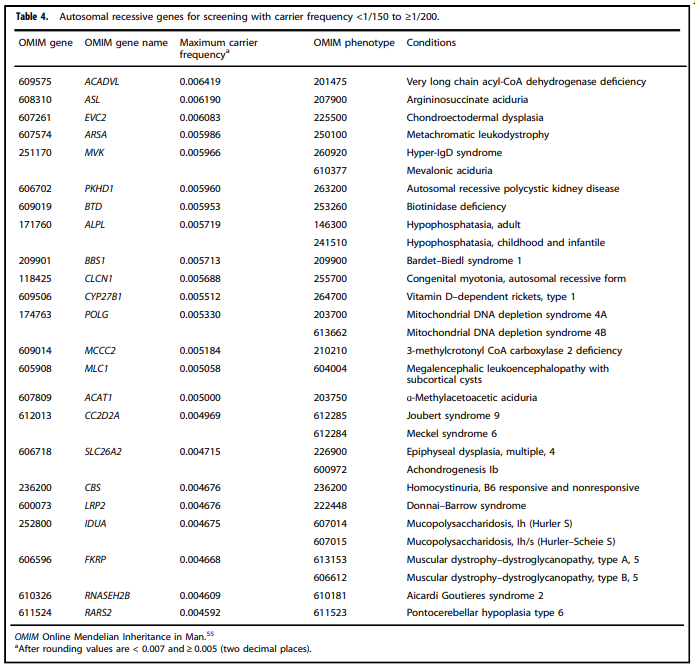
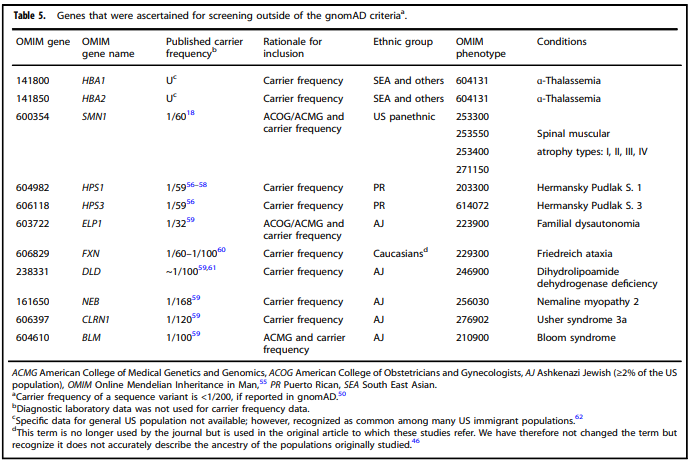
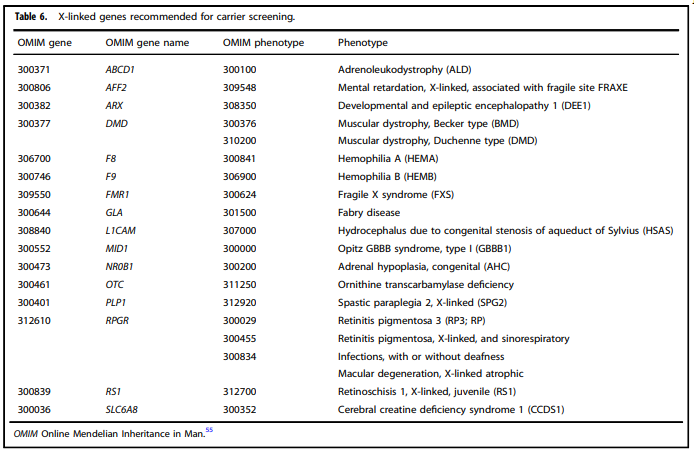
Tables 1 – 6 from (Gregg et al., 2021)
International Society for Prenatal Diagnosis (ISPD), the Society for Maternal Fetal Medicine (SMFM), and the Perinatal Quality Foundation (PQF)
The ISPD, SMFM and PQF published the following guidelines on the use of genome-wide sequencing for fetal diagnosis:
- The use of diagnostic sequencing is currently being introduced for evaluation of fetuses for whom standard diagnostic genetic testing, such as chromosomal microarray analysis (CMA), has already been performed and is uninformative, is offered concurrently according to accepted practice guidelines, or for whom expert genetic opinion determines that standard genetic testing is less optimal than sequencing for the presenting fetal phenotype.
- The routine use of prenatal sequencing as a diagnostic test cannot currently be supported due to insufficient validation data and knowledge about its benefits and pitfalls (ISPD, 2018).
In addition to the joint position statement released in 2018, the IPSD released a guideline in 2020 on the use of cfDNA screening for trisomies in multiple pregnancies:
- “The use of first trimester cfDNA screening for the common autosomal trisomies is appropriate for twin pregnancies due to sufficient evidence showing high detection and low false positive rates with high predictive values. Moderate.”
- “It is preferable for laboratories performing cfDNA testing in multi-fetal pregnancies to take evidence of zygosity into consideration (e.g., chorionicity, sex of the fetuses, embryo transfer history) for the interpretation of both test results and fetal fractions. Moderate.”
- “Screening options for triplet pregnancies are lacking and cfDNA may be a potential option. However, diagnostic testing should always be offered and the limitations of screening tests stressed. Low” (Palomaki et al., 2021).
The Canadian National Rh Working Group and the Society of Obstetricians and Gynaecologists of Canada (SOGC) Genetics Committee
Guidelines were published by a consensus meeting of the Canadian National Rh Working Group in collaboration with the SOGC Genetics committee. The following recommendations were provided:
- “The current optimal management of the D-negative pregnant individual is based on the prediction of the fetal D-blood group by cell-free DNA in maternal plasma with targeted antenatal anti-D prophylaxis. This approach should be adopted in Canada (II-2A).
- While various algorithms of implementation of fetal RHD genotyping have been described, a model positioned in the first trimester appears to be most in alignment with the existing Canadian antenatal anti-D prophylaxis program and should be endorsed (II-2A).
- While the risk of a false-negative result with RHD genotyping is very small and the benefits of knowing the fetal RHD status in terms of compliance with prophylaxis seem to outweigh the risks, the chance of immunization is not zero. Quality control at a laboratory and clinical level should be of utmost priority in program planning (II-3A)” (Johnson et al., 2017).
College of American Pathologists (CAP) Transfusion Medicine Resource Committee (TMRC) Work Group
The following recommendations were given by the CAP RMRC Work Group:
- The Work Group recommends that RHD genotyping be performed whenever a discordant RhD typing result and/or a serological weak D phenotype is detected in patients, including pregnant individuals, newborns, and potential transfusion recipients. It is anticipated that the immediate benefit will be fewer unnecessary injections of RhIG and increased availability of RhD-negative RBCs for transfusion.
- Other than RHD genotypes weak D type 1, 2, or 3, the Work Group recommends that individuals with a serological weak D phenotype receive conventional prophylaxis with RhIG, including postpartum RhIG if the newborn is RhD-positive or has a serological weak D phenotype (Sandler et al., 2015).
Royal College of Obstetricians and Gynaecologists (RCOG)
The RCOG have given the following recommendation for prenatal and fetal genotyping: “Non-invasive fetal genotyping using maternal blood is now possible for D, C, c, E, e and K antigens. This should be performed in the first instance for the relevant antigen when maternal red cell antibodies are present” (C recommendation) (RCOG, 2014).
Table of Terminology
|
Term |
Definition |
|
ACMG |
American College of Medical Genetics and Genomics |
|
ACOG |
American College of Obstetricians and Gynecologists |
|
ADA |
American Diabetes Association |
|
CAP |
College of American Pathologists |
|
cfDNA |
Cell-free deoxyribonucleic acid |
|
CMA |
Chromosomal microarray |
|
CNVs |
Copy number variants |
|
DMD |
Dystrophin |
|
DNA |
Deoxyribonucleic acid |
|
FRAXE |
Fragile site, folic acid type, rare, Fra(X)(Q28) E |
|
GJB6 |
Gap junction protein |
|
HDFN |
Hemolytic disease of the fetus and newborn |
|
ISPD |
International Society for Prenatal Diagnosis |
|
MMS |
Microdeletion/microduplication syndromes |
|
NGS |
Next generation sequencing |
|
NIPS-Plus |
Expanded non-invasive prenatal screening |
|
NIPT |
Non-invasive prenatal testing |
|
NT |
Nuchal translucency |
|
PPVs |
Positive predictive values |
|
PQF |
Perinatal quality foundation |
|
RBC |
Red blood cells |
|
RCOG |
Royal College of Obstetricians and Gynaecologists |
|
RHD |
Rh blood group D antigen |
|
SMA |
Spinal muscular atrophy |
|
SMFM |
Society for Maternal Fetal Medicine |
|
SMN1 |
Survival of motor neuron 1 |
|
SNP |
Single nucleotide polymorphism |
|
TMRC |
Transfusion Medicine Resource Committee |
References:
- ACOG. (2016a). Committee Opinion No.682: Microarrays and Next-Generation Sequencing Technology: The Use of Advanced Genetic Diagnostic Tools in Obstetrics and Gynecology. Obstet Gynecol, 128(6), e262-e268. https://doi.org/10.1097/aog.0000000000001817
- ACOG. (2016b). Prenatal Diagnostic Testing for Genetic Disorders. https://s3.amazonaws.com/cdn.smfm.org/publications/223/download-f5260f3bc6686c15e4780f8100c74448.pdf
- ACOG. (2017a). Carrier Screening in the Age of Genomic Medicine. https://www.acog.org/-/media/Committee-Opinions/Committee-on-Genetics/co690.pdf?dmc=1&ts=20170328T2033175160
- ACOG. (2017b). Practice Bulletin No. 181: Prevention of Rh D Alloimmunization. https://journals.lww.com/greenjournal/fulltext/2017/08000/Practice_Bulletin_No__181__Prevention_of_Rh_D.54.aspx
- ACOG. (2019). Hereditary Cancer Syndromes and Risk Assessment: ACOG COMMITTEE OPINION, Number 793. Obstet Gynecol, 134(6), e143-e149. https://doi.org/10.1097/AOG.0000000000003562
- ACOG. (2020). Carrier Screening in the Age of Genomic Medicine. Retrieved 2/4/21 from https://www.acog.org/clinical/clinical-guidance/committee-opinion/articles/2017/03/carrier-screening-in-the-age-of-genomic-medicine
- ACOG. (2021). Consumer Testing for Disease Risk: ACOG Committee Opinion, Number 816. Obstet Gynecol, 137(1), e1-e6. https://doi.org/10.1097/AOG.0000000000004200
- Biro, O., Rigo, J., Jr., & Nagy, B. (2020). Noninvasive prenatal testing for congenital heart disease - cell-free nucleic acid and protein biomarkers in maternal blood. J Matern Fetal Neonatal Med, 33(6), 1044-1050. https://doi.org/10.1080/14767058.2018.1508437
- Calhoun, D. (2020, 3/23/2020). Postnatal diagnosis and management of hemolytic disease of the fetus and newborn. Retrieved 2/1/2021 from https://www.uptodate.com/contents/postnatal-diagnosis-and-management-of-hemolytic-disease-of-the-fetus-and-newborn?topicRef=6773&source=see_link
- Clausen, F. B., Steffensen, R., Christiansen, M., Rudby, M., Jakobsen, M. A., Jakobsen, T. R., Krog, G. R., Madsen, R. D., Nielsen, K. R., Rieneck, K., Sprogoe, U., Homburg, K. M., Baech, J., Dziegiel, M. H., & Grunnet, N. (2014). Routine noninvasive prenatal screening for fetal RHD in plasma of RhD-negative pregnant women-2 years of screening experience from Denmark. Prenat Diagn, 34(10), 1000-1005. https://doi.org/10.1002/pd.4419
- Daniels, G., Finning, K., Martin, P., & Summers, J. (2007). Fetal RhD genotyping: a more efficient use of anti-D immunoglobulin. Transfus Clin Biol, 14(6), 568-571. https://doi.org/10.1016/j.tracli.2008.03.007
- Darlington, M., Carbonne, B., Mailloux, A., Brossard, Y., Levy-Mozziconacci, A., Cortey, A., Maoulida, H., Simon, T., Rousseau, A., & Durand-Zaleski, I. (2018). Effectiveness and costs of non-invasive foetal RHD genotyping in rhesus-D negative mothers: a French multicentric two-arm study of 850 women. BMC Pregnancy Childbirth, 18(1), 496. https://doi.org/10.1186/s12884-018-2114-5
- de Haas, M., Thurik, F. F., van der Ploeg, C. P., Veldhuisen, B., Hirschberg, H., Soussan, A. A., Woortmeijer, H., Abbink, F., Page-Christiaens, G. C., Scheffer, P. G., & Ellen van der Schoot, C. (2016). Sensitivity of fetal RHD screening for safe guidance of targeted anti-D immunoglobulin prophylaxis: prospective cohort study of a nationwide programme in the Netherlands. Bmj, 355, i5789. https://doi.org/10.1136/bmj.i5789
- de Jong, A., Maya, I., & van Lith, J. M. (2015). Prenatal screening: current practice, new developments, ethical challenges. Bioethics, 29(1), 1-8. https://doi.org/10.1111/bioe.12123
- Dondorp, W., de Wert, G., Bombard, Y., Bianchi, D. W., Bergmann, C., Borry, P., Chitty, L. S., Fellmann, F., Forzano, F., Hall, A., Henneman, L., Howard, H. C., Lucassen, A., Ormond, K., Peterlin, B., Radojkovic, D., Rogowski, W., Soller, M., Tibben, A., . . . American Society of Human, G. (2015). Non-invasive prenatal testing for aneuploidy and beyond: challenges of responsible innovation in prenatal screening. European journal of human genetics : EJHG, 23(11), 1438-1450. https://doi.org/10.1038/ejhg.2015.57
- Finning, K., Martin, P., Summers, J., Massey, E., Poole, G., & Daniels, G. (2008). Effect of high throughput RHD typing of fetal DNA in maternal plasma on use of anti-RhD immunoglobulin in RhD negative pregnant women: prospective feasibility study. Bmj, 336(7648), 816-818. https://doi.org/10.1136/bmj.39518.463206.25
- Gil, M. M., Akolekar, R., Quezada, M. S., Bregant, B., & Nicolaides, K. H. (2014). Analysis of cell-free DNA in maternal blood in screening for aneuploidies: meta-analysis. Fetal Diagn Ther, 35(3), 156-173. https://doi.org/10.1159/000358326
- Grant, A., & Mohide, P. (1982). Screening and diagnostic tests in antenatal care. Effectiveness and satisfaction in antenatal care, 22-59. https://books.google.com/books?hl=en&lr=&id=fVH-JYbe2isC&oi=fnd&pg=PA22&dq=screening+versus+diagnostic+tests&ots=WXVxt6ALwT&sig=DUy8K33sGYU72yPEjPHIyTT3ppA#v=onepage&q=screening %20versus%20diagnostic%20tests&f=false
- Gregg, A. R., Aarabi, M., Klugman, S., Leach, N. T., Bashford, M. T., Goldwaser, T., Chen, E., Sparks, T. N., Reddi, H. V., Rajkovic, A., Dungan, J. S., Practice, A. P., & Guidelines, C. (2021). Screening for autosomal recessive and X-linked conditions during pregnancy and preconception: a practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med, 23(10), 1793-1806. https://doi.org/10.1038/s41436-021-01203-z
- Gregg, A. R., Skotko, B. G., Benkendorf, J. L., Monaghan, K. G., Bajaj, K., Best, R. G., Klugman, S., & Watson, M. S. (2016). Noninvasive prenatal screening for fetal aneuploidy, 2016 update: a position statement of the American College of Medical Genetics and Genomics. Genet Med, 18(10), 1056-1065. https://doi.org/10.1038/gim.2016.97
- ISPD. (2018). Joint Position Statement from the International Society for Prenatal Diagnosis (ISPD), the Society for Maternal Fetal Medicine (SMFM), and the Perinatal Quality Foundation (PQF) on the use of genome-wide sequencing for fetal diagnosis. Prenat Diagn, 38(1), 6-9. https://doi.org/10.1002/pd.5195
- Johnson, J. A., MacDonald, K., Clarke, G., & Skoll, A. (2017). No. 343-Routine Non-invasive Prenatal Prediction of Fetal RHD Genotype in Canada: The Time is Here. J Obstet Gynaecol Can, 39(5), 366-373. https://doi.org/10.1016/j.jogc.2016.12.006
- Kent, J., Farrell, A. M., & Soothill, P. (2014). Routine administration of Anti-D: the ethical case for offering pregnant women fetal RHD genotyping and a review of policy and practice. BMC Pregnancy Childbirth, 14, 87. https://doi.org/10.1186/1471-2393-14-87
- LabCorp. (2020). Inheritest. https://www.integratedgenetics.com/patients/pre-pregnancy/inheritest
- Liang, D., Cram, D. S., Tan, H., Linpeng, S., Liu, Y., Sun, H., Zhang, Y., Tian, F., Zhu, H., Xu, M., Wang, H., Yu, F., & Wu, L. (2019). Clinical utility of noninvasive prenatal screening for expanded chromosome disease syndromes. Genetics in Medicine, 21(9), 1998-2006. https://doi.org/10.1038/s41436-019-0467-4
- Lockwood, C. J., & Magriples, U. (2020, 12/9/2020). Prenatal care: Initial assessment. Wolters Kluwer. Retrieved 02/01/2021 from https://www.uptodate.com/contents/prenatal-care-initial-assessment
- Mackie, F. L., Hemming, K., Allen, S., Morris, R. K., & Kilby, M. D. (2017). The accuracy of cell-free fetal DNA-based non-invasive prenatal testing in singleton pregnancies: a systematic review and bivariate meta-analysis. Bjog, 124(1), 32-46. https://doi.org/10.1111/1471-0528.14050
- Manfroi, S., Calisesi, C., Fagiani, P., Gabriele, A., Lodi, G., Nucci, S., Pelliconi, S., Righini, L., & Randi, V. (2018). Prenatal non-invasive foetal RHD genotyping: diagnostic accuracy of a test as a guide for appropriate administration of antenatal anti-D immunoprophylaxis. Blood Transfus, 16(6), 514-524. https://doi.org/10.2450/2018.0270-17
- Martinez-Payo, C., Bada-Bosch, I., Martinez-Moya, M., & Perez-Medina, T. (2018). Clinical results after the implementation of cell-free fetal DNA detection in maternal plasma. J Obstet Gynaecol Res, 44(8), 1369-1376. https://doi.org/10.1111/jog.13672
- Migliorini, S., Saccone, G., Silvestro, F., Massaro, G., Arduino, B., D'Alessandro, P., Petti, M. T., Paino, J. A. C., Guida, M., Locci, M., & Zullo, F. (2020). First-trimester screening based on cell-free DNA vs combined screening: A randomized clinical trial on women's experience. Prenat Diagn, 40(11), 1482-1488. https://doi.org/10.1002/pd.5800
- Miller, D. (2020, 7/9/2020). Use of chromosomal microarray in obstetrics. Wolters Kluwer. Retrieved 02/01/2021 from https://www.uptodate.com/contents/use-of-chromosomal-microarray-in-obstetrics
- Moise, K. (2020, 12/16/2020). Prevention of RhD alloimmunization in pregnancy. Retrieved 2/2/2021 from https://www.uptodate.com/contents/prevention-of-rhd-alloimmunization-in-pregnancy
- Palomaki, G. E., Chiu, R. W. K., Pertile, M. D., Sistermans, E. A., Yaron, Y., Vermeesch, J. R., Vora, N. L., Best, R. G., & Wilkins-Haug, L. (2021). International Society for Prenatal Diagnosis Position Statement: cell free (cf)DNA screening for Down syndrome in multiple pregnancies. Prenat Diagn, 41(10), 1222-1232. https://doi.org/10.1002/pd.5832
- Persico, N., Boito, S., Ischia, B., Cordisco, A., De Robertis, V., Fabietti, I., Periti, E., Volpe, P., Fedele, L., & Rembouskos, G. J. P. D. V. (2016). Cell‐free DNA testing in the maternal blood in high‐risk pregnancies after first‐trimester combined screening. Prenatal Diagnosis, 36(3), 232-236.
- RCOG. (2014). The Management of Women with Red Cell Antibodies during Pregnancy. https://www.rcog.org.uk/globalassets/documents/guidelines/rbc_gtg65.pdf
- Rink, B., Romero, S., Biggio, J., Saller, D., Giardine, R., & ACOG. (2017). Committee Opinion No. 691: Carrier Screening for Genetic Conditions. Obstet Gynecol, 129(3), e41-e55. https://doi.org/10.1097/aog.0000000000001952
- Romero, S., Rink, B., Biggio, J., Saller, D., & ACOG. (2017). Committee Opinion No. 690: Carrier Screening in the Age of Genomic Medicine. Obstet Gynecol, 129(3), e35-e40. https://doi.org/10.1097/aog.0000000000001951
- Runkel, B., Bein, G., Sieben, W., Sow, D., Polus, S., & Fleer, D. (2020). Targeted antenatal anti-D prophylaxis for RhD-negative pregnant women: a systematic review. BMC Pregnancy Childbirth, 20(1), 83. https://doi.org/10.1186/s12884-020-2742-4
- Sandler, S. G., Flegel, W. A., Westhoff, C. M., Denomme, G. A., Delaney, M., Keller, M. A., Johnson, S. T., Katz, L., Queenan, J. T., Vassallo, R. R., & Simon, C. D. (2015). It's time to phase in RHD genotyping for patients with a serologic weak D phenotype. College of American Pathologists Transfusion Medicine Resource Committee Work Group. Transfusion, 55(3), 680-689. https://doi.org/10.1111/trf.12941
- Vora, N., Ralston, S., & ACOG. (2018). ACOG Technology Assessment in Obstetrics and Gynecology No. 14: Modern Genetics in Obstetrics and Gynecology. Obstet Gynecol, 132(3), e143-e168. https://doi.org/10.1097/aog.0000000000002831
- Witkop, C., & ACOG. (2018). ACOG Committee Opinion No. 727: Cascade Testing: Testing Women for Known Hereditary Genetic Mutations Associated With Cancer. Obstet Gynecol, 131(1), e31-e34. https://doi.org/10.1097/aog.0000000000002457
- Yang, H., Llewellyn, A., Walker, R., Harden, M., Saramago, P., Griffin, S., & Simmonds, M. (2019). High-throughput, non-invasive prenatal testing for fetal rhesus D status in RhD-negative women: a systematic review and meta-analysis. BMC Med, 17(1), 37. https://doi.org/10.1186/s12916-019-1254-4
- Yesilcinar, I., & Guvenc, G. (2021). Counselling and education for prenatal screening and diagnostic tests for pregnant women: Randomized controlled trial. Int J Nurs Pract, 27(5), e13000. https://doi.org/10.1111/ijn.13000
- Zhu, X., Chen, M., Wang, H., Guo, Y., Chau, M. H. K., Yan, H., Cao, Y., Kwok, Y. K. Y., Chen, J., Hui, A. S. Y., Zhang, R., Meng, Z., Zhu, Y., Leung, T. Y., Xiong, L., Kong, X., & Choy, K. W. (2020). Clinical utility of expanded noninvasive prenatal screening and chromosomal microarray analysis in high risk pregnancies. Ultrasound Obstet Gynecol. https://doi.org/10.1002/uog.22021
Coding Section
| Code |
Number |
Code Description |
| CPT |
81171 |
AFF2 (AF4/FMR2 family, member 2 [FMR2]) (e.g., fragile X mental retardation 2 [FRAXE]) gene analysis; evaluation to detect abnormal (eg, expanded) alleles |
|
81172 |
AFF2 (AF4/FMR2 family, member 2 [FMR2]) (e.g., fragile X mental retardation 2 [FRAXE]) gene analysis; characterization of alleles (e.g., expanded size and methylation status) |
|
|
81200 |
ASPA (aspartoacylase) (e.g., Canavan disease) gene analysis, common variants (e.g., E285A, Y231X) |
|
|
81209 |
BLM (Bloom syndrome, RecQ helicase-like) (e.g., Bloom syndrome) gene analysis, 2281del6ins7 variant |
|
|
81241 |
F5 (coagulation factor V) (e.g., hereditary hypercoagulability) gene analysis, Leiden variant |
|
|
81242 |
FANCC (Fanconi anemia, complementation group C) (e.g., Fanconi anemia, type C) gene analysis, common variant (e.g., IVS4+4A>T) |
|
|
81243 |
FMR1 (fragile X mental retardation 1) (e.g., fragile X mental retardation) gene analysis; evaluation to detect abnormal (e.g., expanded) alleles |
|
|
81244 |
FMR1 (fragile X mental retardation 1) (e.g., fragile X mental retardation) gene analysis; characterization of alleles (e.g., expanded size and promoter methylation status) |
|
|
81251 |
GBA (glucosidase, beta, acid) (e.g., Gaucher disease) gene analysis, common variants (e.g., N370S, 84GG, L444P, IVS2+1G>A) |
|
|
81255 |
HEXA (hexosaminidase A [alpha polypeptide]) (e.g., Tay-Sachs disease) gene analysis, common variants (eg, 1278insTATC, 1421+1G>C, G269S) |
|
|
81257 |
HBA1/HBA2 (alpha globin 1 and alpha globin 2) (e.g., alpha thalassemia, Hb Bart hydrops fetalis syndrome, HbH disease), gene analysis; common deletions or variant (e.g., Southeast Asian, Thai, Filipino, Mediterranean, alpha3.7, alpha4.2, alpha20.5, Constant Spring) |
|
|
81260 |
IKBKAP (inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase complex-associated protein) (e.g., familial dysautonomia) gene analysis, common variants (e.g., 2507+6T>C, R696P) |
|
|
81290 |
MCOLN1 (mucolipin 1) (e.g., Mucolipidosis, type IV) gene analysis, common variants (e.g., IVS3-2A>G, del6.4kb) |
|
|
81329 |
SMN1 (survival of motor neuron 1, telomeric) (e.g., spinal muscular atrophy) gene analysis; dosage/deletion analysis (e.g., carrier testing), includes SMN2 (survival of motor neuron 2, centromeric) analysis, if performed |
|
|
81330 |
SMPD1(sphingomyelin phosphodiesterase 1, acid lysosomal) (e.g., Niemann-Pick disease, Type A) gene analysis, common variants (e.g., R496L, L302P, fsP330) |
|
|
81400 |
Molecular pathology procedure, Level 1 (e.g., identification of single germline variant [e.g., SNP] by techniques such as restriction enzyme digestion or melt curve analysis) |
|
|
81401 |
Molecular pathology procedure, Level 2 (e.g., 2 – 10 SNPs, 1 methylated variant, or 1 somatic variant [typically using nonsequencing target variant analysis], or detection of a dynamic mutation disorder/triplet repeat) |
|
|
81403 |
Molecular pathology procedure, Level 4 (e.g., analysis of single exon by DNA sequence analysis, analysis of > 10 amplicons using multiplex PCR in 2 or more independent reactions, mutation scanning or duplication/deletion variants of 2 – 5 exons) |
|
|
81404 |
Molecular pathology procedure, Level 5 (e.g., analysis of 2 – 5 exons by DNA sequence analysis, mutation scanning or duplication/deletion variants of 6 – 10 exons, or characterization of a dynamic mutation disorder/triplet repeat by Southern blot analysis) |
|
|
81405 |
Molecular pathology procedure, Level 6 (e.g., analysis of 6 – 10 exons by DNA sequence analysis, mutation scanning or duplication/deletion variants of 11 – 25 exons, regionally targeted cytogenomic array analysis) |
|
|
81406 |
Molecular pathology procedure, Level 7 (e.g., analysis of 11 – 25 exons by DNA sequence analysis, mutation scanning or duplication/deletion variants of 26 – 50 exons, cytogenomic array analysis for neoplasia) |
|
|
81412 |
Ashkenazi Jewish associated disorders (e.g., Bloom syndrome, Canavan disease, cystic fibrosis, familial dysautonomia, Fanconi anemia group C, Gaucher disease, Tay-Sachs disease), genomic sequence analysis panel, must include sequencing of at least 9 genes, including ASPA, BLM, CFTR, FANCC, GBA, HEXA, IKBKAP, MCOLN1, and SMPD1 |
|
|
81443 |
Genetic testing for severe inherited conditions (e.g., cystic fibrosis, Ashkenazi Jewish-associated disorders [e.g., Bloom syndrome, Canavan disease, Fanconi anemia type C, mucolipidosis type VI, Gaucher disease, Tay-Sachs disease], beta hemoglobinopathies, phenylketonuria, galactosemia), genomic sequence analysis panel, must include sequencing of at least 15 genes (e.g., ACADM, ARSA, ASPA, ATP7B, BCKDHA, BCKDHB, BLM, CFTR, DHCR7, FANCC, G6PC, GAA, GALT, GBA, GBE1, HBB, HEXA, IKBKAP, MCOLN1, PAH) |
|
| 0400U (effective 07/01/2023) | Obstetrics (expanded carrier screening), 145 genes by next-generation sequencing, fragment analysis and multiplex ligation-dependent probe amplification, DNA, reported as carrier positive or negative | |
|
S3845 |
Genetic testing for alpha-thalassemia |
|
|
S3846 |
Genetic testing for hemoglobin E beta-thalassemia |
|
|
S3849 |
Genetic testing for niemann-pick disease |
Procedure and diagnosis codes on Medical Policy documents are included only as a general reference tool for each policy. They may not be all-inclusive.
This medical policy was developed through consideration of peer-reviewed medical literature generally recognized by the relevant medical community, U.S. FDA approval status, nationally accepted standards of medical practice and accepted standards of medical practice in this community, Blue Cross Blue Shield Association technology assessment program (TEC) and other nonaffiliated technology evaluation centers, reference to federal regulations, other plan medical policies, and accredited national guidelines.
"Current Procedural Terminology © American Medical Association. All Rights Reserved"
History From 2022 Forward