
Testing of Homocysteine Metabolism-Related Conditions - CAM 162
Description
Homocystinuria is a metabolic condition in which the body is unable to properly process certain amino acids, resulting in an abnormal accumulation of homocysteine and its metabolites in the blood and urine (NIH, 2018). Homocystinuria is primarily due to genetic causes; however, homocystinuria can also be due to non-genetic causes, including severe deficiency of vitamin B12, also known as cobalamin (Mudd et al., 2000).
Regulatory Status
A search of the FDA Device database on Nov. 1, 2020 for “homocysteine” yielded 30 results. Additionally, many labs have developed specific tests that they must validate and perform in house. These laboratory-developed tests (LDTs) are regulated by the Centers for Medicare & Medicaid Services (CMS) as high-complexity tests under the Clinical Laboratory Improvement Amendments of 1988 (CLIA ’88). As an LDT, the U.S. Food and Drug Administration has not approved or cleared this test; however, FDA clearance or approval is not currently required for clinical use.
On May 13, 2011, the FDA approved the Invader MTHFR 677 created by Hologic, Inc. The Invader MTHFR 677 is an in-vitro diagnostic test intended for the detection and genotyping of a single point mutation (C to T at position 677) of the human 5,10-methylenetetrahydrofolate reductase (MTHFR) gene in isolated genomic DNA obtained from whole blood Potassium EDTA samples from patients with suspected thrombophilia.
On April 25, 2011, the FDA approved the Invader MTHFR 1298 created by Hologic, Inc. The Invader MTHFR 1298 test is an in vitro diagnostic test intended for the detection and genotyping of a single point mutation (A to C at position 1298) of the human 5,10-methylenetetrahydrofo late reductase (MTHFR) gene in isolated genomic DNA obtained from whole blood potassium EDTA samples from patients with suspected thrombophilia.
On April 22, 2010, the FDA approved the eSensor Thrombophilia Risk Test on XT-8 System created by Osmetech Molecular Diagnostics. The MTHFR-specific portion is as follows: The eSensor MTHFR Genotyping Test is an in-vitro diagnostic for the detection and genotyping of point mutations (C to T at position 677) and (A to C at position 1298) of the human 5,10-methylenetetrahydrofo late reductase (MTHFR) gene in isolated genomic DNA obtained from whole blood samples. The test is intended to be used on the eSensor XT-8 System.
On October 11, 2007, the FDA approved the Verigene System created by Nanosphere Inc. The MTHFR-specific portion is as follows: The Verigene MTHFR Nucleic Acid Test is an in vitro diagnostic for the detection and genotyping of a single point mutation (C to T at position 677) of the human 5,10-methylenetetrahydrofolate reductase gene (MTHFR) in patients with suspected thrombophilia, from isolated genomic DNA obtained from whole blood samples. The test is intended to be used on the Verigene System (FDA, 2020).
Policy
- Genetic counseling is recommended prior to genetic testing for homocystinuria.
- For individuals with signs and symptoms consistent with classical homocystinuria, genetic testing of CBS is considered MEDICALLY NECESSARY
- Genetic testing for variants of MTHFR known to cause homocystinuria is considered MEDICALLY NECESSARY for either of the following conditions:
- For symptomatic individuals patients that have tested negative for classical homocystinuria (due to CBS deficiency)
- For individuals with a first-degree relative (see Note 1) positive for known variants of MTHFR that cause homocystinuria
- Newborn screening for homocysteine-related conditions is considered MEDICALLY NECESSARY in any of the following situations:
- Screening for classic homocystinuria due to CBS deficiency by performing quantitative plasma amino acids analysis and/or plasma or urine total homocysteine analysis
- Screening for homocystinuria in dried blood spots
- Screening for hypermethioninemia in dried blood spots
- When the initial screening test result exceeds the cut-off level of methionine, a repeat dried blood specimen submitted to the newborn screening program, or a quantitative plasma amino acid analysis and analysis of plasma total homocysteine is considered MEDICALLY NECESSARY.
- For the diagnosis of phenotype variants of classic homocystinuria due to CBS deficiency, the pyridoxine (B6) challenge test is considered MEDICALLY NECESSARY.
- For individuals over 18 years of age with homocystinuria suspected to be caused by CBS deficiency and for monitoring therapy in those with confirmed CBS deficiency, total homocysteine testing in plasma is considered MEDICALLY NECESSARY.
- Plasma free homocysteine testing is considered NOT MEDICALLY NECESSARY.
The following does not meet coverage criteria due to a lack of available published scientific literature confirming that the test(s) is/are required and beneficial for the diagnosis and treatment of a patient’s illness.
- Genetic testing for MTR, MTRR, and MMADHC genes is considered NOT MEDICALLY NECESSARY.
NOTES:
Note 1: First-degree relatives include parents, full siblings, and children of the individual.
Table of Terminology
|
Term |
Definition |
|
3-MST |
3-mercaptopyruvate |
|
ACMG |
American College of Medical Genetics |
|
cblC |
Homocystinuria type C |
|
cblD |
Cobalamin D |
|
cblD-Hcy |
Cobalamin D homocysteine |
|
CblE |
Methylcobalamin type E |
|
cblF |
Cobalamin F |
|
cblG |
Methylcobalamin type G |
|
cblJ |
Methylcobalamin type J |
|
CBS |
Cystathionine β-synthase |
|
CLIA ’88 |
Clinical Laboratory Improvement Amendments of 1988 |
|
CMS |
Centers for Medicare & Medicaid |
|
CT |
Computed tomography |
|
HT |
Heterozygous state |
|
CTH |
Cystathionine |
|
DNA |
Deoxyribonucleic acid |
|
EDTA |
Chelating agentin deoxyribose nucleic acid extraction |
|
E-HOD |
European Network and Registry for Homocystinuria and Methylation Defects |
|
FDA |
Food and Drug Administration |
|
FPIA |
Fluorescence polarization immunoassay |
|
GCLC |
Glutamylcysteine |
|
GC-MS |
Gas chromatography-mass spectrometry |
|
Hcy |
Homocysteine |
|
HHS |
The U.S. Department of Health and Human Services |
|
HPLC |
High performance liquid chromatography |
|
LC-MS |
Liquid chromatography-mass spectrometry |
|
LC-MS-MS |
Liquid chromatography tandem mass spectrometry |
|
LDTs |
Laboratory-developed tests |
|
MENA |
Middle East and North Africa |
|
Met |
Methionine |
|
MMA |
Methylmalonic acid |
|
MMADHC |
Methylmalonic aciduria and homocystinuria type D protein |
|
MRI |
Magnetic resonance imaging |
|
MTHFR |
Methylene tetrahydrofolate reductase |
|
MTR |
Methionine synthase |
|
MTRR |
Methionine synthase reductase |
|
NIH |
National Institutes of Health |
|
Phe |
Phenylalanine |
|
SAM |
S-adenosyl methionine |
|
tHcy |
Total homocysteine |
|
TT |
Homozygous state |
|
UPLC-MS/MS |
Ultra performance liquid chromatography-tandem mass spectrometry |
Rationale
Homocysteine (Hcy), a naturally occurring intermediary amino acid, is involved in multiple metabolic pathways, including the transsulfuration pathway as well as methionine (Met) metabolism. Classical homocystinuria, which results in an accumulation of Hcy and its metabolites in the blood and urine, is due to genetic mutations in cystathionine-β-synthase (CBS). CBS is the enzyme responsible for the rate-limiting step of the transsulfuration pathway and is dependent on pyridoxine (vitamin B6) (Zhu et al., 2018). If this enzyme is blocked, the transsulfuration of Hcy and the accumulation of both Hcy and Met will be limited, as Met concentration is enhanced by remethylation. The disruption of the Met metabolic pathway, as shown in Figure 1 below (Zhu et al., 2018), prevents Hcy from being used properly; this creates in a buildup of Hcy and toxic by-products in the blood, with excess Hcy excreted in urine (Mazaheri et al., 2017).
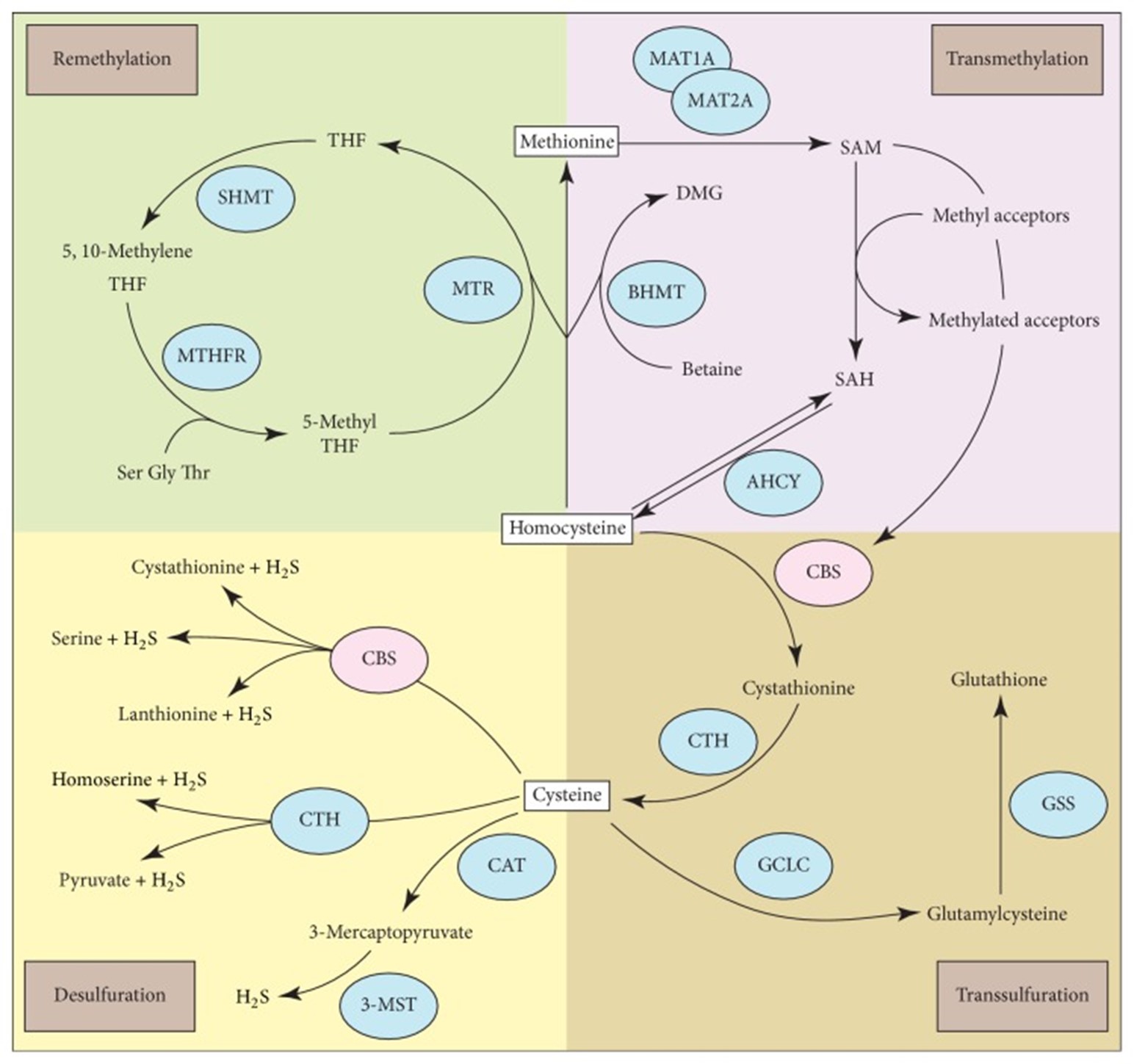
Figure 1: Homocysteine is a common metabolite linked to multiple metabolic pathways, including methionine/S-adenosyl methionine (SAM) metabolism, transsulfuration, and desulfuration. Figure taken from (Zhu et al., 2018).
Homocystinuria due to CBS deficiency can cause eye problems, skeletal abnormalities, an increased risk for blood clots, and developmental delay. Homocystinuria may also generate white matter abnormalities in the brain, potentially mimicking other disorders such as leukoencephalopathy when imaged with computed tomography (CT) and magnetic resonance imaging (MRI) scans (Ismayilova et al., 2019; Li et al., 2018).
The exact incidence of homocystinuria due to CBS deficiency is unknown. In 1985, the incidence was estimated to be around 1:344,000 worldwide (Mudd et al., 1985). However, the National Institutes of Health (NIH) is now estimating these rates to be much higher, around 1:150,000 worldwide, 1:200,000 – 300,000 in the United States, and 1:1,800 in Qatar (NIH, 2018). In European populations, incidence rates have been predicted by molecular epidemiological studies to be between 1:6,400 and 1:20,500 (Gaustadnes et al., 1999; Janosík et al., 2009). Higher prevalence in the MENA (Middle East and North Africa) region could be attributed to high consanguinity in those communities (Al-Sadeq & Nasrallah, 2020). Infants with homocystinuria due to CBS deficiency are asymptomatic at birth, with symptoms slowly developing if left untreated. However, these symptoms are highly variable. Some affected patients may exhibit mild symptoms of the disorder while others may develop potentially life-threatening complications. Depending on the population affected and type of CBS gene mutation, symptoms can be as severe as ectopia lentis, Marfanoid features, mental retardation, idiopathic infertility, osteoporosis, and severe premature atherosclerosis (Al-Sadeq & Nasrallah, 2020; Rosenson et al., 2020). The phenotype of these patients mainly relates to pyridoxine-responsiveness: Pyridoxine treatment responders exhibit a milder phenotype and a later onset than pyridoxine treatment nonresponders (Abbott et al., 1987; Mudd et al., 1985). Early detection and treatment is important in preventing or reducing the severity of the disorder. Screening for homocystinuria is frequently incorporated into state newborn screening programs (Rose & Dolan, 2012). While a newborn blood spot specimen for hypermethioninemia will detect homocystinuria due to CBS deficiency in some, not all affected individuals will be detected by this test (Sacharow et al., 2004).
According to Sacharow et al. (2004), the biochemical features of homocystinuria include:
- Markedly increased concentrations of total homocysteine, plasma homocysteine, homocysteine-cysteine mixed disulfide and methionine.
- Increased concentration of homocysteine in urine.
- Reduced CBS enzyme activity.
Classical biochemical findings establishing the diagnosis are summarized in the following table titled: Cardinal Biochemical Findings that Establish the Diagnosis of Homocystinuria (Sacharow et al., 2004).
|
Analyte |
Specimen |
Expected Findings |
||
|
Neonate with |
Untreated older individual |
Control |
||
|
Total homocysteine (tHcy) |
Plasma |
50 to > 100 µmol/L |
> 100 µmol/L |
< 15 µmol/L |
|
Methionine (on amino acid analysis) |
Plasma |
200 – 1500 µmol/L |
> 50 µmol/L |
10 – 40 µmol/L |
Homocystinuria due to genetic causes is inherited in an autosomal recessive pattern. Many different forms of homocystinuria can occur and signs and symptoms vary depending on the gene mutation. CBS gene mutations cause the most common form of homocystinuria. This mutation is referred to as “classic” homocystinuria or CBS deficiency. Other gene mutations that can result in homocystinuria include MTHFR, MTR, MTRR, and MMADHC. The MTHFR, MTR, and MTRR genes all revolve around the remethylation pathway of Hcy, while the MMADHC gene plays a role in Vitamin B12 metabolism (Froese et al., 2015; Wang et al., 2016).
Homocystinuria may also be associated with a diagnosis of methylmalonic acidemia, when the body cannot efficiently break down specific fats or proteins, leading to a methylmalonic acid buildup in the blood. Methylmalonic aciduria and homocystinuria type C (cblC) is characterized by a vitamin B12 disorder initiated by a mutation in the MMACHC gene; symptoms of this disorder fall into several categories, including thromboembolic and neurological issues such as cognitive and psychiatric episodes (Collison et al., 2015).
Analytical Validity
This concentration of total homocysteine (tHcy) in blood plasma is the primary clinical analyte measured to diagnose homocystinuria. A study using liquid chromatography-mass spectrometry (LC-MS) calculated limits of detection (0.06 µmol/L) and quantification (0.6 µmol/Lu) of tHcy (Nelson et al., 2003). Another study using gas chromatography-mass spectrometry (GC-MS) found a detection limit of 0.4 µmol/L as well as intra- and inter-run variations of 5% and 8%, respectively. Furthermore, this method was found to compare well with the LC-MS-MS method; the GC-MS method had a mean difference of -0.4 µmol compared to the LC-MS-MS method (Belkhiria et al., 2007). Fluorescence polarization immunoassay (FPIA) was found to compare favorably to the high performance liquid chromatography (HPLC) and MS approaches as well (5% imprecision with -2% to 3% bias) so it is a practical option if the more precise approaches are not available; unfortunately, this study only measured levels up to 45 µmol/L, whereas severe homocystinuria can exceed 100 µmol/L (Nexo et al., 2000).
More recently, Concepción-Alvarez et al. (2016) have validated a method to quantify Hcy in plasma samples via HPLC. Hcy levels were measured in a total of 46 patients and the authors found that HPLC was able to “identify and quantify Hcy without interferences” and that the identified detection limit was 3.12 μM and quantification limit 6.25 μM (Concepción-Alvarez et al., 2016). This research has provided further validation for Hcy plasma testing in ailments such as homocystinuria where this amino acid is increased.
For the detection of Hcy-related conditions, methylmalonic acid and tHcy are commonly measured in both plasma samples and dried blood spots. Using ultra performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS), de Sain-van der Velden et al. (2015) recently compared methylmalonic acid and tHcy levels collected either from a dried blood spot or from plasma concentration testing methods to determine which is the more efficient and accurate method. The authors note that the plasma assay performed better than the dried blood spot testing method in most areas, but that dried blood spot testing was superior for tHcy stability. Furthermore, a strong correlation of tHcy was found in both testing methods, (y = 0.46 ± 1.12 (r(2) = 0.91)), leading to the authors suggestion that tHcy testing in plasma can be replaced by tHcy in dried blood spots (de Sain-van der Velden et al., 2015).
Clinical Utility and Validity
A diagnosis of classic homocystinuria (caused by CBS deficiency) is established by measurement of tHcy. The normal level is < 15 µMol/L, whereas a newborn with homocystinuria is expected to measure at > 50 µMol/L and an older, untreated individual will likely measure at > 100 µMol/L (Sacharow et al., 2004). A measurement of Met in plasma can corroborate a diagnosis, as the metabolic pathway involves a buildup of Met in addition to the buildup of Hcy (Sacharow et al., 2004). While free Hcy composes about 15 – 25% of tHcy levels, separate free Hcy testing is unnecessary: tHcy measurement already includes all forms of Hcy (Rosenson et al., 2020).
The detection of biallelic pathogenic variants in CBS can substantiate a diagnosis of classic homocystinuria (Sacharow et al., 2004). There are two phenotypic variants in homocystinuria, both caused by CBS: B6-responsive and B6-non-responsive homocystinuria. The pyridoxine (B6) challenge test is performed to determine the variant and if vitamin B6 therapy will be beneficial (Sacharow et al., 2004). Testing for homocystinuria usually involves biochemical testing in urine and/or genetic testing for known mutations. Genetic testing can be done using a single gene or multi-gene panel which may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests. Homocystinuria typically involves CBS deficiency and while the activity of the CBS enzyme could be performed in cultured fibroblasts when genetic tests are inconclusive, enzymatic testing for CBS deficiency is no longer available in USA (Sacharow et al., 2004).
Methylene tetrahydrofolate reductase (MTHFR) mutations are of interest in homocystinuria. The MTHFR enzyme catalyzes the reduction of 5,10-methylenetetrahydrofolate to 5,10-methyltetrahydrofolate, the methyl donor for the conversion of Hcy to Met. Failure of this enzyme ( < 20% of normal levels) leads to increased Hcy and Met, as well as the production of other symptoms associated with homocystinuria (Long & Goldblatt, 2016). The two most common mutations in the MTHFR gene are 677T (changed from a C nucleotide) and 1298C (changed from an A). Both mutations can be heterozygotic or homozygotic, and both can lead to loss of enzymatic function (Gonzales et al., 2017). The 677T mutation is more severe, as in the homozygous state (TT) it results in up to 70% loss of enzymatic function, compared to only a 35% loss of function in the heterozygous state (CT) (Frosst et al., 1995). The 1298C mutation results in a loss of enzymatic function; 30% and 15% for its homozygous and heterozygous forms, respectively (Weisberg et al., 1998).
However, it is possible that dietary factors (notably low levels of folate or Vitamin B12) influence tHcy levels more than genetic factors. A study covering 452 young adults found tHcy variance to have a 9% total genetic contribution (i.e. genetic polymorphisms) compared to a 35% contribution from dietary factors. The only polymorphism found to have a significant effect on tHcy levels was the 677T mutation, which interacted with low folate levels to produce a high tHcy phenotype. Compared to the authors earlier studies of genetic influence on tHcy levels, the younger cohort’s genetic contribution on tHcy levels was measured out to be higher than the older cohort’s (9% compared to 7% for the older cohort). Furthermore, the authors suggest that genetic influence on tHcy levels are more pronounced during early life and environmental factors are more influential as time passes (Gaughan et al., 2001; Harmon et al., 1999; Kluijtmans et al., 2003).
Another study conducted by Gales et al. (2018) found that focal epilepsy presentation in the context of adult or adolescent onset could result from a mutation leading to MTHFR deficiency. It is critical that this mutation found in homocystinuria be detected early for treatment, as the neuropsychiatric syndrome could be easily treated with a combination of vitamin B9, vitamin B12, and betaine (Gales et al., 2018).
A novel newborn screening method has been developed by researchers: a two-tier algorithm using a methionine (Met) to phenylalanine (Phe) ratio. Data from 125,047 neonates was utilized to determine this accuracy of this method (Okun et al., 2017). It was reported that “Met to Phe ratio was found to be more effective for first sieve than Met, sorting out nearly 90% of normal samples. Only 10% of the samples would have to be processed by second-tier measurement of Hcy in dried blood spots” (Okun et al., 2017). This novel testing method resulted in 100% sensitivity and specificity for classical homocystinuria newborn screening (Okun et al., 2017).
The United States Department of Health and Human Services
The Secretary of the U.S. Department of HHS has developed a recommended uniform screening panel for every universal newborn screening program; the amino acid disorder homocystinuria is recommended as a core condition for newborn screening, and the organic acid condition methylmalonic acidemia with homocystinuria is recommended as a secondary screening condition. Methylmalonic acidemia due to methylmalonyl-CoA mutase or cobalamin disorders is included as a core condition as well (HHS, 2018).
American College of Medical Genetics (ACMG)
ACMG recommends quantitative testing of plasma amino acids to determine increased levels of Hcy and Met; classical homocystinuria is characterized by increases in both Hcy and Met, while increased Met may be indicative of other disorders (ACMG, 2010). Also, plasma Hcy analysis will show increased Hcy in classical homocystinuria and normal or only slightly increased Hcy in the other disorders. Urine Hcy will be significantly increased in classical homocystinuria (ACMG, 2010).
In the Confirmatory Algorithms for Met, ACMG indicates that increased Met and increased tHcy are indicative of homocystinuria due to CBS deficiency (ACMG, 2021).
European Network and Registry for Homocystinuria and Methylation Defects (E-HOD)
In 2015, a project by the E-HOD released the Newborn Screening for Homocystinurias and Methylation Disorders: Systematic Review and Proposed Guidelines. In this guideline, authors recommend newborn screening for CBS deficiency by detecting elevated Met, methionine-to-phenylalanine ratio, and/or tHcy in dried blood spots. Specificity is increased by analyzing tHcy as a second-tier marker and calculating Met/tHcy ratio is also suggested (Huemer et al., 2015)
Newborn screening for the cblD-Hcy, CblE, and cblG defects, and for MTHFR deficiency could be possible by measuring Met and methionine-to-phenylalanine ratio in dried blood spots followed by analysis of tHcy as a second-tier marker. However, it is stated that the efficacy and feasibility of screening for these disorders is largely unknown (Huemer et al., 2015).
As a part of E-HOD, the Guidelines for Diagnosis and Management of The Cobalamin-Related Remethylation Disorders cblC, cblD, cblE, cblF, cblG, cblJ and MTHFR Deficiency were released in 2017. Huemer et al. (2017) “strongly recommend measuring plasma total homocysteine in any patient presenting with the combination of neurological and/or visual and/or haematological symptoms, subacute spinal cord degeneration, atypical haemolytic uraemic syndrome or unexplained vascular thrombosis.” For a “valid, timely laboratory diagnosis,” the authors also add:
- “We strongly recommend that investigations in patients with a suspected remethylation disorder should start with the measurement of total homocysteine in blood. We recommend the blood sample for tHcy to be centrifuged within an hour and kept at +4° or frozen until analysis. Immunoassays or chromatographic methods are suitable for tHcy measurement. (Quality of the evidence: moderate)
- We strongly recommend against measuring free homocysteine instead of total homocysteine. (Quality of the evidence: moderate)
- We strongly recommend that in the case of high total homocysteine, plasma and urine samples for determination of MMA, methionine, folate and vitamin B12 are to be obtained before treatment is started. (Quality of the evidence: moderate)” (Huemer et al., 2017).
Another guideline written as a part of E-HOD provides practical guides to recognition, diagnosis and management of CBS deficiency. The guideline presented 41 separate recommendations based on a literature review by the Guideline Development Group and the authors admitted that the quality of the identified data was poor and many of their recommendations were grade D; however, the highest recommendation was given to measuring the plasma total homocysteine concentrations in any patient whose signs and symptoms strongly suggest the diagnosis (Morris et al., 2017).
For the biochemical diagnosis, a tHcy test is recommended as “the frontline test” for the diagnosis of CBS deficiency. Plasma free Hcy is only detectable at tHcy concentrations above 50 – 60 µmol/L; its measurement is not particularly sensitive or even reproducible and is, therefore, not recommended. Untreated patients with a CBS deficiency typically have tHcy concentrations above 100 μmol/L and a diagnosis is likely if an elevated tHcy is found along with high or borderline high plasma Met concentrations. Further information such as low plasma cystathionine concentration or increased Met:Cystathionine ratio can support a diagnosis. Finally, tHcy measurement using dried blood spots can be done if plasma processing is not possible.
E-HOD recommends confirming CBS deficiency by measuring cystathionine synthase activity in fibroblasts or plasma and/or by mutation analysis of CBS gene. The gold standard for confirming CBS deficiency is determination of cystathionine production of Hcy and serine in cultured fibroblasts. Either the enzyme or DNA can be analyzed and if one method does not confirm a diagnosis, the other method should be done. The grade of this recommendation is B – C.
Despite technical pitfalls of DNA testing, E-HOD recommends a molecular genetic analysis of the CBS gene for the confirmation of CBS deficiency and for carrier and prenatal testing (grade B). For the prenatal diagnosis, the molecular analysis is a preferred technique during the first trimester of pregnancy. If the mutations are known in the family, enzyme analysis can be performed in cultured amniocytes, but not in chorionic villi. Preimplantation analysis could also be done (grade C – D).
For newborn screening, it is recommended to increase specificity of Met testing by using tHcy as a second marker and calculating Met/tHcy ratio (grade C). Several other metabolic disorders can cause an increased Met concentration and the exact sensitivity of detecting Met in newborns with a CBS deficiency is unknown. Although the median Met concentration of CBS deficient patients is far greater than the median of a healthy neonate (103 µmol/L compared to 20 µmol/L), individual Met values may still vary.
Screening for family members at risk is recommended by measuring tHcy but molecular genetic testing may also be utilized in exceptional cases (grade D).
Monitoring of tHcy, amino acids, folate and vitamin B12 is recommended in all patients during therapy. The frequency of the monitoring is variable on a case-by-case basis (due to severity, treatment plan, age, etc.). The targeted concentration ranges for total plasma homocysteine are proposed to be < 50 µmol/L in pyridoxine-responsive patients and at < 11 µmol/L free homocysteine (about 120 µmol/L total homocysteine) in pyridoxine-unresponsive patients (Morris et al., 2017).
References
- Abbott, M. H., Folstein, S. E., Abbey, H., & Pyeritz, R. E. (1987). Psychiatric manifestations of homocystinuria due to cystathionine beta-synthase deficiency: prevalence, natural history, and relationship to neurologic impairment and vitamin B6-responsiveness. Am J Med Genet, 26(4), 959-969. https://doi.org/1v0.1002/ajmg.1320260427
- ACMG. (2010). Newborn Screening ACT Sheet [Increased Methionine] Homocystinuria (CBS Deficiency) https://www.ncbi.nlm.nih.gov/books/NBK55827/
- ACMG. (2021). Methionine Elevated or Decreased. American College of Medical Genetics and Genomics. Retrieved 11/7/2022 from http://www.acmg.net/PDFLibrary/Methionine-Algorithm.pdf
- Al-Sadeq, D. W., & Nasrallah, G. K. (2020). The Spectrum of Mutations of Homocystinuria in the MENA Region. Genes (Basel), 11(3). https://doi.org/10.3390/genes11030330
- Belkhiria, M. N., Ducros, V., Harzallah, K., Jarraya, F., Cordonnier, D., Favier, A., & Achour, A. (2007). [Evaluation of plasmatic homocysteine determination by gas chromatography-mass spectrometry]. Ann Biol Clin (Paris), 65(4), 393-398. (Evaluation d'un transfert de methode : dosage de l'homocysteine plasmatique par chromatographie en phase gazeuse couplee a la spectrometrie de masse.)
- Collison, F. T., Xie, Y. A., Gambin, T., Jhangiani, S., Muzny, D., Gibbs, R., Lupski, J. R., Fishman, G. A., & Allikmets, R. (2015). Whole Exome Sequencing Identifies an Adult-Onset Case of Methylmalonic Aciduria and Homocystinuria Type C (cblC) with Non-Syndromic Bull's Eye Maculopathy. Ophthalmic Genet, 36(3), 270-275. https://doi.org/10.3109/13816810.2015.1010736
- Concepción-Alvarez, A., Camayd-Viera, I., & Nuevas-Paz, L. (2016). Validation of an HPLC method for total homocysteine quantification in plasma. Revista del Laboratorio Clinico, 9(2), 40-47. https://www.sciencedirect.com/science/article/pii/S1888400816300022#!
- de Sain-van der Velden, M. G. M., van der Ham, M., Jans, J. J., Visser, G., van Hasselt, P. M., Prinsen, H., & Verhoeven-Duif, N. M. (2015). Suitability of methylmalonic acid and total homocysteine analysis in dried bloodspots. Anal Chim Acta, 853, 435-441. https://doi.org/10.1016/j.aca.2014.10.043
- FDA. (2007, October 11, 2007). Nanosphere 510(k) Summary. Retrieved October 21, 2021 from https://www.accessdata.fda.gov/cdrh_docs/pdf7/K070597.pdf
- FDA. (2010, March 24, 2010). eSensor Thrombophila Risk Test on XT-8 System. Retrieved October 21, 2021 from https://www.accessdata.fda.gov/cdrh_docs/pdf9/K093974.pdf
- FDA. (2011a, May 13, 2011). Invader MTHFR 677 510(k) Summary. Retrieved October 21, 2021 from https://www.accessdata.fda.gov/cdrh_docs/pdf10/K100987.pdf
- FDA. (2011b, April 25, 2011). Invader MTHFR 1298 510(k) Summary. Retrieved October 21, 2021 from https://www.accessdata.fda.gov/cdrh_docs/pdf10/K100496.pdf
- Froese, D. S., Kopec, J., Fitzpatrick, F., Schuller, M., McCorvie, T. J., Chalk, R., Plessl, T., Fettelschoss, V., Fowler, B., Baumgartner, M. R., & Yue, W. W. (2015). Structural Insights into the MMACHC-MMADHC Protein Complex Involved in Vitamin B12 Trafficking. J Biol Chem, 290(49), 29167-29177. https://doi.org/10.1074/jbc.M115.683268
- Frosst, P., Blom, H. J., Milos, R., Goyette, P., Sheppard, C. A., Matthews, R. G., Boers, G. J., den Heijer, M., Kluijtmans, L. A., van den Heuvel, L. P., & et al. (1995). A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet, 10(1), 111-113. https://doi.org/10.1038/ng0595-111
- Gales, A., Masingue, M., Millecamps, S., Giraudier, S., Grosliere, L., Adam, C., Salim, C., Navarro, V., & Nadjar, Y. (2018). Adolescence/adult onset MTHFR deficiency may manifest as isolated and treatable distinct neuro-psychiatric syndromes. Orphanet J Rare Dis, 13(1), 29. https://doi.org/10.1186/s13023-018-0767-9
- Gaughan, D. J., Kluijtmans, L. A., Barbaux, S., McMaster, D., Young, I. S., Yarnell, J. W., Evans, A., & Whitehead, A. S. (2001). The methionine synthase reductase (MTRR) A66G polymorphism is a novel genetic determinant of plasma homocysteine concentrations. Atherosclerosis, 157(2), 451-456.
- Gaustadnes, M., Ingerslev, J., & Rütiger, N. (1999). Prevalence of Congenital Homocystinuria in Denmark. New England Journal of Medicine, 340(19), 1513-1513. https://doi.org/10.1056/NEJM199905133401915
- Gonzales, M. C., Yu, P., & Shiao, S. P. K. (2017). MTHFR Gene Polymorphism-Mutations and Air Pollution as Risk Factors for Breast Cancer: A Metaprediction Study. Nursing research, 66(2), 152-163. https://doi.org/10.1097/NNR.0000000000000206
- Harmon, D. L., Shields, D. C., Woodside, J. V., McMaster, D., Yarnell, J. W., Young, I. S., Peng, K., Shane, B., Evans, A. E., & Whitehead, A. S. (1999). Methionine synthase D919G polymorphism is a significant but modest determinant of circulating homocysteine concentrations. Genet Epidemiol, 17(4), 298-309. https://doi.org/10.1002/(sici)1098-2272(199911)17:4<298::Aid-gepi5>3.0.Co;2-v
- HHS. (2018, 02/2019). Recommended Uniform Screening Panel. https://www.hrsa.gov/advisory-committees/heritable-disorders/rusp/index.html
- Huemer, M., Diodato, D., Schwahn, B., Schiff, M., Bandeira, A., Benoist, J. F., Burlina, A., Cerone, R., Couce, M. L., Garcia-Cazorla, A., la Marca, G., Pasquini, E., Vilarinho, L., Weisfeld-Adams, J. D., Kožich, V., Blom, H., Baumgartner, M. R., & Dionisi-Vici, C. (2017). Guidelines for diagnosis and management of the cobalamin-related remethylation disorders cblC, cblD, cblE, cblF, cblG, cblJ and MTHFR deficiency. J Inherit Metab Dis, 40(1), 21-48. https://doi.org/10.1007/s10545-016-9991-4
- Huemer, M., Kožich, V., Rinaldo, P., Baumgartner, M. R., Merinero, B., Pasquini, E., Ribes, A., & Blom, H. J. (2015). Newborn screening for homocystinurias and methylation disorders: systematic review and proposed guidelines. Journal of inherited metabolic disease, 38(6), 1007-1019. https://doi.org/10.1007/s10545-015-9830-z
- Ismayilova, N., MacKinnon, A. D., Mundy, H., & Fallon, P. (2019). Reversible Cerebral White Matter Abnormalities in Homocystinuria. JIMD Rep, 44, 115-119. https://doi.org/10.1007/8904_2018_135
- Janosík, M., Sokolová, J., Janosíková, B., Krijt, J., Klatovská, V., & Kozich, V. (2009). Birth prevalence of homocystinuria in Central Europe: frequency and pathogenicity of mutation c.1105C>T (p.R369C) in the cystathionine beta-synthase gene. The Journal of pediatrics, 154(3), 431-437. https://doi.org/10.1016/j.jpeds.2008.09.015
- Kluijtmans, L. A., Young, I. S., Boreham, C. A., Murray, L., McMaster, D., McNulty, H., Strain, J. J., McPartlin, J., Scott, J. M., & Whitehead, A. S. (2003). Genetic and nutritional factors contributing to hyperhomocysteinemia in young adults. Blood, 101(7), 2483-2488. https://doi.org/10.1182/blood.V101.7.2483
- Li, Barshop, Feigenbaum, & Khanna. (2018). Brain Magnetic Resonance Imaging Findings in Poorly Controlled Homocystinuria. J Radiol Case Rep. https://www.ncbi.nlm.nih.gov/pubmed/29875981
- Long, S., & Goldblatt, J. (2016). MTHFR genetic testing: Controversy and clinical implications. Australian Family Physician, 45, 237-240. http://www.racgp.org.au/afp/2016/april/mthfr-genetic-testing-controversy-and-clinical-implications/
- Mazaheri, Mostofizadeh, & Hashemipour. (2017). Homocystinuria with Stroke and Positive Familial History. Adv Biomed Res. https://www.ncbi.nlm.nih.gov/pubmed/29279830
- Morris, A. A. M., Kožich, V., Santra, S., Andria, G., Ben-Omran, T. I. M., Chakrapani, A. B., Crushell, E., Henderson, M. J., Hochuli, M., Huemer, M., Janssen, M. C. H., Maillot, F., Mayne, P. D., McNulty, J., Morrison, T. M., Ogier, H., O’Sullivan, S., Pavlíková, M., de Almeida, I. T., . . . Chapman, K. A. J. J. o. I. M. D. (2017). Guidelines for the diagnosis and management of cystathionine beta-synthase deficiency [journal article]. 40(1), 49-74. https://doi.org/10.1007/s10545-016-9979-0
- Mudd, S. H., Finkelstein, J. D., Refsum, H., Ueland, P. M., Malinow, M. R., Lentz, S. R., Jacobsen, D. W., Brattstrom, L., Wilcken, B., Wilcken, D. E., Blom, H. J., Stabler, S. P., Allen, R. H., Selhub, J., & Rosenberg, I. H. (2000). Homocysteine and its disulfide derivatives: a suggested consensus terminology. Arterioscler Thromb Vasc Biol, 20(7), 1704-1706.
- Mudd, S. H., Skovby, F., Levy, H. L., Pettigrew, K. D., Wilcken, B., Pyeritz, R. E., Andria, G., Boers, G. H., Bromberg, I. L., Cerone, R., & et al. (1985). The natural history of homocystinuria due to cystathionine beta-synthase deficiency. Am J Hum Genet, 37(1), 1-31.
- Nelson, B. C., Pfeiffer, C. M., Sniegoski, L. T., & Satterfield, M. B. (2003). Development and evaluation of an isotope dilution LC/MS method for the determination of total homocysteine in human plasma. Anal Chem, 75(4), 775-784.
- Nexo, E., Engbaek, F., Ueland, P. M., Westby, C., O’Gorman, P., Johnston, C., Kase, B. F., Guttormsen, A. B., Alfheim, I., McPartlin, J., Smith, D., Møller, J., Rasmussen, K., Clarke, R., Scott, J. M., & Refsum, H. (2000). Evaluation of Novel Assays in Clinical Chemistry: Quantification of Plasma Total Homocysteine. Clinical Chemistry, 46(8), 1150. http://clinchem.aaccjnls.org/content/46/8/1150.abstract
- NIH. (2018). Homocystinuria due to CBS deficiency. https://rarediseases.info.nih.gov/diseases/6667/homocystinuria-due-to-cbs-deficiency
- Okun, J. G., Gan-Schreier, H., Ben-Omran, T., Schmidt, K. V., Fang-Hoffmann, J., Gramer, G., Abdoh, G., Shahbeck, N., Al Rifai, H., Al Khal, A. L., Haege, G., Chiang, C. C., Kasper, D. C., Wilcken, B., Burgard, P., & Hoffmann, G. F. (2017). Newborn Screening for Vitamin B6 Non-responsive Classical Homocystinuria: Systematical Evaluation of a Two-Tier Strategy. JIMD Rep, 32, 87-94. https://doi.org/10.1007/8904_2016_556
- Rose, N. C., & Dolan, S. M. (2012). Newborn screening and the obstetrician. Obstetrics and gynecology, 120(4), 908-917. https://doi.org/10.1097/AOG.0b013e31826b2f03
- Rosenson, R. S., Smith, C. C., & Bauer, K. A. (2020, October 26). Overview of homocysteine. Wolters Kluwer. Retrieved November 1 from https://www.uptodate.com/contents/overview-of-homocysteine
- Sacharow, S. J., Picker, J. D., & Levy, H. L. (2004). Homocystinuria Caused by Cystathionine Beta-Synthase Deficiency. In M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, & A. Amemiya (Eds.), GeneReviews((R)). University of Washington, Seattle. https://www.ncbi.nlm.nih.gov/pubmed/20301697
- Wang, W., Jiao, X. H., Wang, X. P., Sun, X. Y., & Dong, C. (2016). MTR, MTRR, and MTHFR Gene Polymorphisms and Susceptibility to Nonsyndromic Cleft Lip With or Without Cleft Palate. Genet Test Mol Biomarkers, 20(6), 297-303. https://doi.org/10.1089/gtmb.2015.0186
- Weisberg, I., Tran, P., Christensen, B., Sibani, S., & Rozen, R. (1998). A second genetic polymorphism in methylenetetrahydrofolate reductase (MTHFR) associated with decreased enzyme activity. Mol Genet Metab, 64(3), 169-172. https://doi.org/10.1006/mgme.1998.2714
- Zhu, H., Blake, S., Chan, K. T., Pearson, R. B., & Kang, J. (2018). Cystathionine beta-Synthase in Physiology and Cancer. Biomed Res Int, 2018, 3205125. https://doi.org/10.1155/2018/3205125
Coding Section
|
Code Number |
Code Description |
|
81291 |
MTHFR (5,10-methylenetetrahydrofolate reductase) (e.g., hereditary hypercoagulability) gene analysis, common variants (e.g., 677T, 1298C) |
|
81401 |
Molecular pathology procedure, Level 2 (e.g., 2 – 10 SNPs, 1 methylated variant, or 1 somatic variant [typically using nonsequencing target variant analysis], or detection of a dynamic mutation disorder/triplet repeat) |
|
81406 |
Molecular pathology procedure, Level 7 (e.g., analysis of 11 – 25 exons by DNA sequence analysis, mutation scanning or duplication/deletion variants of 26 – 50 exons, cytogenomic array analysis for neoplasia) |
|
81479 |
Unlisted molecular pathology procedure |
|
82136 |
Amino acids, 2 to 5 amino acids, quantitative, each specimen |
|
82139 |
Amino acids, 6 or more amino acids, quantitative, each specimen |
|
82615 |
Cystine and homocystine, urine, qualitative |
|
83090 |
Homocysteine |
|
83921 |
Organic acid, single, quantitative |
|
84207 |
Pyridoxal phosphate (Vitamin B-6) |
|
96040 |
Medical genetics and genetic counseling services, each 30 minutes face-to-face with patient/family |
|
S0265 |
Genetic counseling, under physician supervision, each 15 minutes |
Procedure and diagnosis codes on Medical Policy documents are included only as a general reference tool for each policy. They may not be all-inclusive.
This medical policy was developed through consideration of peer-reviewed medical literature generally recognized by the relevant medical community, U.S. FDA approval status, nationally accepted standards of medical practice and accepted standards of medical practice in this community, Blue Cross Blue Shield Association technology assessment program (TEC) and other non-affiliated technology evaluation centers, reference to federal regulations, other plan medical policies and accredited national guidelines.
"Current Procedural Terminology© American Medical Association. All Rights Reserved"
History From 2017 Forward
| 01/30/2023 | Annual review, updating policy for clarity and consistency. Adding note/guideline describing first degree relatives. Also updating description, rationale and references. |
|
01/12/2022 |
Annual review, no change to policy intent. Updating rationale and references. |
|
01/08/2021 |
Annual review, medical necessity criteria updated to include age and therapy verbiage. Also reformatting policy for clarity including description, background, regulatory status, rationale and references. |
|
01/06/2020 |
Annual review, no change to policy intent. Updating coding. |
|
09/24/2019 |
Updated coding. No other changes made. |
|
01/08/2019 |
Annual review, adding medical necessity statement regarding newborn testing for hypermethioninemia in dried blood spots. Also adding limited coverage for 81291/ MTHFR testing. Updating coding. |
|
06/26/2018 |
Interim Review. Updated diagnosis coding. No other changes made |
|
01/30/2018 |
Annual review, expanding medical necessity coverage for some newborn issues. Adding criteria for pyridoxine challenge testing. Adding criteria for testing for suspected CBS deficiency. No other changes. |
|
04/26/2017 |
Interim review to align with Avalon quarterly schedule. Updated category to Laboratory. |
|
04/12/2017 |
Corrected formatting in policy statement. |
|
03/23/2017 |
New Policy |